Breaking the Barrier: Advanced Strategies to Overcome Catalytic Scaling Relationships for Enhanced Efficiency
This article provides a comprehensive analysis of scaling relationships, a fundamental limitation in heterogeneous catalysis where the binding energies of reaction intermediates are linearly correlated, capping catalytic performance.
Breaking the Barrier: Advanced Strategies to Overcome Catalytic Scaling Relationships for Enhanced Efficiency
Abstract
This article provides a comprehensive analysis of scaling relationships, a fundamental limitation in heterogeneous catalysis where the binding energies of reaction intermediates are linearly correlated, capping catalytic performance. Tailored for researchers and scientists, we explore the theoretical foundations of these relationships, detail innovative strategies to circumvent them—including dynamic site regulation and dual-site mechanisms—and discuss experimental validation through operando techniques. By synthesizing foundational knowledge with cutting-edge methodological advances, this review serves as a critical resource for the rational design of next-generation high-efficiency catalysts, with profound implications for energy conversion and sustainable chemical processes.
The Scaling Relationship Dilemma: Foundations, Origins, and Catalytic Limitations
Defining Linear Scaling Relationships (LSRs) in Catalysis
Frequently Asked Questions (FAQs)
1. What are Linear Scaling Relationships (LSRs) in catalysis? Linear Scaling Relationships (LSRs) are observed correlations where the adsorption energies of different reaction intermediates on a catalyst surface are linearly related [1]. This means that the binding strength of one intermediate (e.g., *OH) can predict the binding strength of another (e.g., *OOH). These relationships arise because the adsorption energies of chemically similar intermediates (like *OH, *O, and *OOH in the oxygen evolution reaction) are correlated and often cannot be adjusted independently on a single active site [1].
2. Why are LSRs a fundamental limitation in catalytic design? LSRs impose an intrinsic limitation on a catalyst's maximal achievable performance and/or selectivity [1]. In multi-step reactions, it becomes thermodynamically challenging to optimally adjust the adsorption energy for every intermediate simultaneously. A catalyst that binds one intermediate strongly will often bind another too weakly, creating a compromise that limits the overall catalytic activity. This is often visualized on a theoretical overpotential volcano plot, where the summit represents the best possible activity under these constraints.
3. What strategies exist to overcome LSRs? Conventional strategies involve engineering heterogeneity into the catalyst to selectively stabilize certain intermediates over others. This can be done by confining intermediates within nanoscopic channels or creating multifunctional surfaces and interfacial sites [1]. An emerging, unconventional paradigm is the dynamic structural regulation of active sites [1]. This approach involves active sites that change their coordination and electronic structure during the catalytic cycle, thereby modulating adsorption energies for different intermediates independently and circumventing the static limitations of LSRs.
4. Can you provide a real-world example of breaking LSRs? A 2025 study demonstrated the breaking of LSRs in the electrochemical oxygen evolution reaction (OER) using a model Ni-Fe₂ molecular catalyst [1]. During the catalytic cycle, dynamic evolution of the Ni-adsorbate coordination, driven by intramolecular proton transfer, altered the electronic structure of the adjacent Fe active center. This dynamic dual-site cooperation simultaneously lowered the free energy change for O–H bond cleavage and O–O bond formation, thereby disrupting the inherent scaling relationship [1].
5. What experimental techniques are used to study LSRs and dynamic sites? Studying dynamic sites requires advanced operando or in situ techniques that can probe the catalyst under working conditions. Key methodologies include:
- Operando X-ray Absorption Fine Structure (XAFS): Used to verify local structural transformations and coordination environments of metal atoms during electrochemical activation and reaction [1].
- Electrokinetic Studies: Analyze reaction kinetics to infer mechanistic pathways and rate-determining steps.
- Density Functional Theory (DFT) & Ab Initio Molecular Dynamics (AIMD) Simulations: Computational methods used to model reaction pathways, free energy changes, and the dynamic evolution of active sites [1].
Troubleshooting Common Experimental Challenges
Problem: Inconsistent catalytic performance when attempting to replicate dynamic catalyst systems.
| Symptom | Potential Cause | Solution |
|---|---|---|
| Low activity and no redox shift in CV | Failed formation of the target molecular complex (e.g., Ni-Fe trimer). | Ensure precise control during in situ electrochemical activation. Verify the purity of the electrolyte (e.g., use Fe-free KOH) and the concentration of intentionally added metal ions (e.g., 1 ppm Fe) [1]. |
| Formation of nanoparticles instead of atomically dispersed sites. | Overly harsh synthesis or activation conditions. | Optimize thermal annealing temperature and atmosphere. Use a support with high defect density (e.g., holey graphene nanomesh) to anchor single atoms and prevent aggregation [1]. |
| Poor reproducibility of operando XAFS data. | Unstable catalyst structure or inconsistent electrochemical conditioning. | Standardize the activation protocol (e.g., using consistent CV cycles, anodic chronopotentiometry). Ensure the electrochemical cell for operando measurements is properly designed to maintain potential control and electrolyte flow. |
Experimental Protocol: Constructing a Dynamic Ni-Fe Molecular Catalyst
This protocol summarizes the methodology for constructing a dynamic Ni-Fe₂ OER catalyst, as reported in recent literature [1].
1. Synthesis of Ni Single-Atom Pre-catalyst (Ni-SAs@GNM)
- Material: Prepare a graphene oxide (GO) aqueous suspension.
- Assembly: Seal the GO suspension in a Ni vessel at 80°C to spontaneously assemble a 3D Ni(OH)₂/graphene hydrogel.
- Drying: Subject the hydrogel to freeze-drying to obtain an aerogel.
- Annealing: Thermally anneal the aerogel at 700°C under an Ar atmosphere. This step reduces Ni(OH)₂ and etches the graphene into a holey graphene nanomesh (GNM).
- Purification: Treat the product with acid to remove nanoparticles, yielding a sample of Ni single atoms trapped on GNM (Ni-SAs@GNM). Confirm the atomic dispersion using aberration-corrected HAADF-STEM.
2. In Situ Electrochemical Activation to Form Ni-Fe Complex
- Electrode Preparation: Load the Ni-SAs@GNM pre-catalyst onto a glassy carbon working electrode.
- Electrolyte Preparation: Use a purified Fe-free 1 M KOH electrolyte with a deliberate addition of a low concentration (e.g., 1 ppm) of Fe ions (present as Fe(OH)₄⁻ anions) [1].
- Activation Procedure: Perform electrochemical activation using Cyclic Voltammetry (CV), typically between 1.1 and 1.65 V vs. RHE. Alternatively, anodic chronopotentiometry or chronoamperometry can be employed. The electrical field drives Fe(OH)₄⁻ to anchor onto Ni sites, forming the active Ni-Fe molecular complex.
The following table summarizes key quantitative data related to the Ni-Fe catalyst system and LSRs.
Table 1: Key Experimental Parameters and Findings from a Study on Breaking LSRs [1]
| Parameter | Value / Description | Significance / Function |
|---|---|---|
| Fe ion concentration | 1 ppm in 1 M KOH | Deliberate addition to form the heteronuclear active site during electrochemical activation. |
| Ni content in pre-catalyst | 0.82 wt% (determined by ICP-OES) | Quantifies the loading of the single-atom pre-catalyst. |
| Specific surface area | 266.9 m² g⁻¹ (for Ni-SAs@GNM) | Indicates a mesoporous structure beneficial for mass transport. |
| Ni/Fe atomic ratio | ~5.2:1 (after activation) | Determined by SXRF and ICP-OES, confirming Fe incorporation. |
| Primary characterization | Operando XAFS, HAADF-STEM, Electrokinetics | Techniques used to identify dynamic structural change and mechanism. |
| Key mechanistic insight | Dynamic Ni-adsorbate coordination alters Fe site electronics | The proposed mechanism for simultaneously optimizing O-H cleavage and O-O formation, thereby breaking the LSR. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Their Functions for Dynamic Catalyst Studies
| Item | Function in the Experiment |
|---|---|
| Graphene Oxide (GO) suspension | Starting material for creating the 3D conductive support structure. |
| Ni Vessel | Serves as a source of Ni ions for the spontaneous formation of Ni(OH)₂ during hydrogel assembly. |
| Purified KOH electrolyte | Provides the alkaline reaction environment; purity is critical to avoid unintended metal contamination. |
| Fe salt (e.g., Fe(NO₃)₃) | Source of Fe ions added at ppm levels to the electrolyte for in situ active site formation. |
| Synchrotron Radiation Source | Enables high-resolution operando XAFS measurements to probe the local structure of atoms under reaction conditions. |
Conceptual Diagrams of LSRs and Breaking Strategies
LSR Limitation in OER
Dynamic Site Regulation
Frequently Asked Questions (FAQs)
Q1: What is the fundamental principle of Bond Order Conservation (BOC) in catalysis?
Bond Order Conservation is a theoretical principle stating that the total bond order between a central atom and its ligands remains constant. When an adsorbate binds to a catalyst surface, the formation of new adsorbate-surface bonds occurs at the expense of the internal bond orders within the adsorbate itself. The sum of these bond orders is conserved. This principle is the foundational origin for the observed scaling relationships in heterogeneous catalysis, which linearly relate the adsorption energies of different reaction intermediates across various catalyst surfaces [2] [3] [4].
Q2: How do scaling relationships limit catalytic performance?
Scaling relationships create a fundamental limitation known as the "catalytic ceiling" or "volcano plot" relationship. Because the adsorption energies of different reaction intermediates are linearly correlated, it becomes impossible to independently optimize the binding strength for all intermediates involved in a reaction. Strengthening the binding of one intermediate inevitably strengthens the binding of others, often stabilizing rate-limiting transition states too much or destabilizing key intermediates. This interdependence places a maximum on the theoretical catalytic activity for a given class of materials, such as transition metals [5].
Q3: What strategies can be used to overcome the limitations of scaling relationships?
Advanced strategies focus on breaking the linear constraints imposed by simple scaling:
- Using BOC-derived Microkinetic Modeling: Applying BOC principles to calculate transition state energies and construct microkinetic models helps identify the specific reaction components that determine overall activity, as demonstrated for NH₃ decomposition [3].
- Designing Integrative Catalytic Pairs (ICPs): These feature spatially adjacent, electronically coupled dual active sites that function cooperatively yet independently. This functional differentiation allows for concerted reactions involving multiple intermediates, moving beyond the limitations of uniform active sites [6].
- Exploring Unconventional Catalytic Environments: Applying electromagnetic fields, using plasma catalysis, or leveraging plasmonic effects can alter the energy landscape and potentially circumvent the constraints of thermal catalysis scaling relationships [7].
Q4: How is the bond order quantified and used in practice?
The bond order parameters (( \lambdai )) for individual bonds are normalized (( \gammai = \lambdai / x{max} )) so that their sum is unity when the central atom satisfies its octet rule or reaches its maximum bond order [4]: [ \sum{i}^{K} \gammai = 1 ] Here, ( K ) is the number of bonds and ( x{max} ) is the maximum number of ligands. This formalism allows for the prediction of adsorption energies for intermediates (e.g., C₂Hₓ species) on different metals based on the adsorption energy of a central atom (e.g., carbon), with a valence parameter ( \gamma(x) = (x{max} - x)/x_{max} ) describing the slope of the scaling relation [4].
Troubleshooting Guides
Issue 1: Poor Predictive Accuracy of Scaling Models
| Symptom | Possible Cause | Solution |
|---|---|---|
| Predicted adsorption energies significantly deviate from experimental or DFT-calculated values. | Model is applied to adsorbates with complex internal bonding (e.g., multiple double/triple bonds) not accounted for in simple AHₓ schemes [4]. | Classify surfaces as "reactive" (π-bond destroying) or "noble" (π-bond preserving) and adjust the internal bond orders of the adsorbate accordingly before applying scaling relations [4]. |
| Model does not account for species-specific differences in key metabolizing enzymes, transporters, or protein binding between different test systems (common in allometric scaling for drug development) [8]. | Incorporate in vitro data (e.g., drug metabolism, plasma protein binding) using In Vitro/In Vivo Extrapolation (IVIVE) or move to more complex Physiologically Based Pharmacokinetic (PBPK) modeling [8]. | |
| Inconsistent transition state energy predictions. | Using linear fits to a set of calculated transition state energies can introduce error [3]. | Apply a BOC-based scheme that requires a limited set of input data to achieve lower errors in transition state energies as a function of simple descriptors [3]. |
Issue 2: Identifying and Modeling Beyond Linear Scaling Relationships
| Symptom | Possible Cause | Solution |
|---|---|---|
| Catalyst optimization hits a performance plateau predicted by a volcano plot. | The catalyst material class (e.g., pure transition metals) is constrained by inherent scaling relationships between key intermediates [5]. | Shift catalyst design strategy. Explore materials with different coordination environments, such as single-atom alloys, oxides, sulfides, or nitrides, which can alter adsorption sites and break simple linear scaling [6] [7]. |
| The catalyst shows poor selectivity in a complex reaction network. | Uniform active sites on single-atom catalysts or simple metals cannot optimally interact with multiple, different intermediates [6]. | Design Integrative Catalytic Pairs (ICPs) or dual-atom sites where spatially adjacent, electronically coupled active sites function cooperatively to handle different reaction steps independently [6]. |
Key Experimental & Computational Protocols
Protocol 1: Establishing a Scaling Relationship for Hydrocarbon Intermediates
This protocol is based on the methodology used to establish scaling relations for C₂Hₓ species on transition metal surfaces [4].
1. Objective: To determine the linear scaling relationship between the adsorption energies of various C₂Hₓ intermediates and the adsorption energy of a central carbon atom across multiple transition metal surfaces.
2. Methodology:
- Computational Setup: Perform Density Functional Theory (DFT) calculations using a plane-wave basis set and ultrasoft pseudopotentials (e.g., with the RPBE functional). Use a plane-wave cut-off energy of 340 eV [4].
- Surface Models: Perform calculations on a representative set of transition metal surfaces, including close-packed surfaces (e.g., fcc(111), hcp(0001), bcc(110)) and stepped surfaces (e.g., fcc(211)) [4].
- Energy Calculations:
- Calculate the adsorption energy (( \Delta E_{M}^{A} )) of a carbon atom on each metal surface
M. - Calculate the adsorption energy (( \Delta E{M}^{AHx} )) for a series of C₂Hₓ intermediates (e.g., x = 0, 1, 2, 3, 4, 5, 6) on the same set of surfaces.
- Calculate the adsorption energy (( \Delta E_{M}^{A} )) of a carbon atom on each metal surface
- Data Analysis:
- For each intermediate C₂Hₓ, plot its adsorption energy on different metals against the adsorption energy of carbon on those same metals.
- Perform a linear regression for each intermediate. The slope of the line is the valence parameter, ( \gamma ).
- The relationship is given by: [ \Delta E{M2}^{AHx} = \Delta E{M1}^{AHx} + \gamma(x)(\Delta E{M2}^{A} - \Delta E{M1}^{A}) ] where ( \gamma(x) = (x{max} - x)/x{max} ), and ( x_{max} ) is the maximum number of hydrogen atoms needed to satisfy the octet rule for the atom forming the bond to the surface [4].
Protocol 2: Applying BOC to Microkinetic Modeling of a Reaction
This protocol outlines how to use BOC to model the kinetics of a catalytic reaction, such as ammonia decomposition [3].
1. Objective: To establish a microkinetic model for the formation of products (e.g., N₂ and H₂ from NH₃) to identify the reaction components that determine catalytic activity.
2. Methodology:
- Energetics Input: Use a BOC-based scheme to calculate the transition state energies and structures of reaction intermediates on a series of transition metal surfaces. This requires only a limited set of input data (e.g., atomic adsorption energies) and is more accurate than simple linear fits [3].
- Model Construction:
- Map the full reaction pathway, identifying all elementary steps (e.g., for NH₃ decomposition: successive dehydrogenation steps and N₂ recombination).
- Use the BOC-derived energies for intermediates and transition states as input parameters for the microkinetic model.
- Simulation & Analysis:
- Run the microkinetic model to simulate reaction rates and product distributions under various conditions (temperature, pressure).
- Analyze the model to identify the rate-determining steps and surface coverages that control the overall activity across different metals. This provides insight for rational catalyst design [3].
Data Presentation
Table 1: Scaling Relation Valence Parameters (γ) for Selected Intermediates
This table summarizes the valence parameter γ for different adsorbate types, which defines their scaling relationship with the atomic adsorbate according to the equation ( \Delta E{M}^{AHx} \propto \gamma \cdot \Delta E_{M}^{A} ) [4].
| Adsorbate Type | Example Intermediates | Valence Parameter (γ) | Key Consideration |
|---|---|---|---|
| Hydrogenated Atoms | CH₃, NH₂, OH | ( \gamma = (x{max} - x)/x{max} ) | The parameter x is the number of H atoms in the intermediate. x_max is 4 for C, 3 for N, and 2 for O [4]. |
| C₂ Species (on noble metals) | C₂Hₓ (with intact π-bonds) | Slope depends on preserved internal multiple bonds. | On noble metal surfaces, internal π-bonds of the hydrocarbon are not broken upon adsorption, affecting the slope [4]. |
| C₂ Species (on reactive metals) | C₂Hₓ (with destroyed π-bonds) | Slope depends on the saturation of the bonding carbon atom. | On reactive metals, the π-system is destroyed, and the scaling is determined by the saturation level of the carbon atom bonded to the surface [4]. |
Table 2: Research Reagent Solutions for Catalysis Studies
| Item / Reagent | Function / Application |
|---|---|
| Density Functional Theory (DFT) Codes (e.g., Dacapo, VASP) | Calculating adsorption energies, transition states, and electronic structures of catalyst surfaces [4]. |
| Transition Metal Surfaces (fcc, hcp, bcc) & Stepped Surfaces (e.g., fcc(211)) | Serving as model systems to study structure-sensitivity and establish scaling relationships across the periodic table [3] [4]. |
| Microkinetic Modeling Software (e.g., Python, MATLAB scripts) | Simulating the overall reaction rate and selectivity based on elementary step energetics to identify performance descriptors [3]. |
| Plasmonic Nanoparticles (e.g., Au, Ag) | Generating strong local electromagnetic fields and hot carriers to drive reactions via unconventional pathways [7]. |
Conceptual Diagrams
Diagram 1: BOC Principle and Scaling Relationship
BOC and Scaling Principle
This diagram illustrates the core principle. During adsorption, new surface bonds (blue) are formed, weakening the adsorbate's internal bonds (yellow). The Bond Order Conservation principle dictates that the total bond order is maintained. This leads to a linear scaling relationship (green-to-red gradient) where the adsorption energies of different intermediates on weak-binding metals predict their energies on strong-binding metals.
Diagram 2: Overcoming Scaling Limitations
Breaking the Scaling Limit
This workflow outlines the problem and solutions. Conventional single-site catalysts (top path) are constrained by linear scaling relationships, leading to performance plateaus. To overcome this, researchers can employ strategies (bottom path) such as designing Integrative Catalytic Pairs (ICPs) with dual sites or using unconventional catalytic environments, which can break the scaling constraints and enhance performance.
Frequently Asked Questions (FAQs)
1. What is a Linear Scaling Relationship (LSR) and how does it limit catalyst activity? In multi-step catalytic reactions like the oxygen evolution reaction (OER), the adsorption energies of different reactive intermediates (such as *OH, *O, and *OOH) are often linearly correlated on conventional single-site catalysts. These Linear Scaling Relationships (LSRs) create a fundamental constraint, making it impossible to independently and optimally adjust the adsorption strength of every intermediate to achieve the maximum theoretical activity [1].
2. How can a Volcano Plot visualize the limitations set by LSRs? A volcano plot is a scatterplot that visualizes the relationship between catalyst activity (often represented as the reaction rate or overpotential) and a descriptor variable (typically the adsorption energy of a key intermediate). The "volcano" shape arises because activity increases as the descriptor energy approaches an optimal value, and then decreases as the energy becomes too strong or too weak. The peak of the volcano represents the maximum activity achievable within the constraints of the LSRs [1].
3. Our experimental data shows points far from the volcano curve. What does this mean? Data points that lie significantly above the predicted volcano curve are highly significant. They indicate that your catalyst system may be successfully circumventing the classic LSRs. This is often achieved through advanced catalyst design strategies, such as creating dynamic active sites or dual-site cooperation, which allow for a more independent adjustment of intermediate binding energies [1].
4. What are the key considerations for creating a statistically sound volcano plot? The two most critical variables are the measure of activity (e.g., log of the turnover frequency) and the descriptor (e.g., adsorption energy). Ensure your statistical thresholds for significance (p-value) and magnitude of change (e.g., log2 fold change) are chosen appropriately for your specific dataset and research question. Incorrect thresholds can misrepresent the number and identity of significant "hits" [9].
5. What strategies exist to break LSRs and reach the top of the volcano? Recent research has demonstrated that LSRs are not absolute barriers. Successful strategies include:
- Dynamic Structural Regulation: Constructing catalysts where the active site dynamically changes its coordination during the catalytic cycle, thereby altering the electronic structure for different reaction steps [1].
- Dual-Site Cooperation: Employing adjacent metal centers that work in concert, where one site participates in the reaction to modulate the electronic structure of the other active site [1].
- Confinement Effects: Using nanoscopic channels or introducing proton acceptors to selectively stabilize certain intermediates over others [1].
Troubleshooting Guide: Common Experimental Issues
| Problem | Possible Cause | Solution |
|---|---|---|
| No significant points on the volcano plot after Differential Gene Expression (DGE) analysis. | Overly stringent statistical thresholds (p-value or fold change). | Re-evaluate threshold choices based on your experimental system. Consider using adjusted p-values to control for false discoveries [9]. |
| Poor separation between significant and non-significant data points. | The chosen descriptor does not effectively correlate with activity for your catalyst system. | Explore alternative descriptor variables that may have a more fundamental relationship with the catalytic activity for your specific reaction. |
| Unexpected clustering of data points in a single region of the plot. | Underlying similarity in the electronic or geometric structure of the tested catalysts. | This can be a valuable finding, indicating a common limitation or shared property across your catalyst library that warrants further investigation. |
| Catalyst performance in experiments does not align with volcano plot predictions. | The assumed reaction mechanism (e.g., Adsorbate Evolution Mechanism) may not be operative. The catalyst may undergo surface reconstruction under operating conditions. | Perform operando characterization (e.g., XAFS) to identify the true active site and mechanism under reaction conditions [1]. |
Experimental Protocol: Generating a Volcano Plot from DGE Data
This protocol outlines the steps to create a basic volcano plot using R, based on differential gene expression results.
Step 1: Environment Setup and Data Import Load the necessary R libraries and import your dataset. The dataset should contain columns for gene identifiers, p-values, and log2 fold change values.
Step 2: Define Significance Thresholds Add a new column to classify each gene (or catalyst) based on your chosen thresholds for statistical significance and magnitude of effect.
Step 3: Construct the Basic Volcano Plot
Use ggplot2 to create the plot, adding threshold lines to guide interpretation.
Step 4: Customize and Export the Plot Refine the plot's appearance by setting a theme and adjusting visual elements. Finally, export the plot for publication.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function / Role in Experiment |
|---|---|
| Graphene Oxide (GO) Suspension | Serves as a precursor for creating a 3D conductive carbon support structure, facilitating the formation of hydrogels and aerogels [1]. |
| Ni Vessel / Ni Salt Precursor | The source of Nickel atoms. Used to construct single-atom pre-catalysts (e.g., Ni-SAs@GNM) which can be dynamically transformed into active sites [1]. |
| Fe Ion Solution (e.g., Fe(OH)₄⁻) | Introduced at ppm levels into the electrolyte for in situ electrochemical doping. Essential for forming heterobinuclear active sites (e.g., Ni-Fe complexes) [1]. |
| Purified KOH Electrolyte | Provides the alkaline reaction environment for OER. Must be purified to be Fe-free to control the incorporation of Fe ions and ensure experimental reproducibility [1]. |
| DGE Results File (.csv) | A comma-separated values file containing the essential data columns: gene/catalyst identifier, p-value, and log2 fold change, which serve as the direct input for volcano plot generation [9]. |
Conceptual Workflow: From Catalyst Design to Overcoming LSRs
The following diagram illustrates the logical workflow and key concepts involved in moving from a traditional catalyst limited by LSRs to an advanced design that can circumvent these relationships.
In the quest for efficient energy conversion technologies, such as water electrolyzers and fuel cells, the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) are pivotal. These reactions are constrained by a fundamental catalytic principle: linear scaling relationships (LSRs). These are linear correlations between the adsorption energies of key oxygenated intermediates—*OH (hydroxyl), *O (oxygen), and *OOH (hydroperoxyl)—on catalyst surfaces. Because these intermediates are chemically similar, their adsorption strengths cannot be adjusted independently [10]. This inherent scaling imposes a thermodynamic overpotential, creating an "activity cliff" that limits the performance of even the most promising catalysts. This technical support article, framed within the broader thesis of overcoming these scaling relationships, provides a practical guide for researchers navigating the experimental challenges in OER and ORR catalyst development.
Frequently Asked Questions (FAQs)
Q1: What are linear scaling relationships (LSRs) and why are they a problem for the Oxygen Evolution Reaction (OER)?
LSRs describe the linear correlations between the adsorption energies of different reaction intermediates on a catalyst surface. In the OER, which follows the Adsorbate Evolution Mechanism (AEM), the key intermediates are *OH, *O, and *OOH [11]. The problem arises because the adsorption energy of *OOH is almost always linearly correlated with the adsorption energy of *OH on conventional single-site catalysts [10] [11]. This correlation means that if a catalyst binds *OH optimally, it will bind *OOH too weakly, or vice-versa. This creates a fundamental thermodynamic limitation, preventing the simultaneous optimization of all reaction steps and capping the maximum achievable activity [11].
Q2: Is the *O vs. *OH scaling relation more important than the *OOH vs. *OH relation for OER trends?
Emerging research suggests that the scaling relation between *O and *OH has been largely overlooked but is critically important for understanding OER activity trends. While the *OOH vs. *OH relationship has been the primary focus in the literature, the *O vs. *OH relationship is equally significant for identifying material motifs and constructing accurate volcano plots to guide catalyst discovery [10]. A comprehensive descriptor approach should consider both relationships to effectively capture catalytic trends.
Q3: Why do my measured ORR activities on platinum show a strong dependence on the electrochemical scan rate?
This is a common experimental challenge rooted in the dynamic nature of the platinum surface. Common half-cell measurements using the rotating disk electrode (RDE) protocol deliver ORR activities that are intrinsically linked to the chosen scan rate. This is because the Pt surface is not static; surface oxygen species (*OH and *O) form and reduce as a function of potential and time. At different scan rates, the surface is in a different state of oxidation, which physically blocks active sites and electronically alters the binding energy of ORR intermediates. Therefore, the measured current is not purely kinetic but is convoluted with these surface processes, making the choice of scan rate somewhat arbitrary from a fundamental perspective [12].
Q4: What are the key strategies for breaking the scaling relationships in OER catalysis?
The most effective strategies involve moving beyond static, single-site catalysts. The primary approach is to engineer heterogeneity to selectively stabilize the *OOH intermediate over *OH. This can be achieved through:
- Dynamic Structural Regulation: Creating active sites that dynamically change their coordination during the catalytic cycle to modulate electronic structure [11].
- Dual-Site Cooperation: Employing multi-functional surfaces or interfaces where different sites cooperate to stabilize different intermediates [11].
- Confinement Effects: Using nanoscopic channels or pockets to confine intermediates.
- Introducing Proton Acceptors: Incorporating functional groups that can act as proton acceptors during the reaction [11].
Troubleshooting Guide
| Problem | Potential Cause | Solution |
|---|---|---|
| Inconsistent ORR kinetics on Pt | Scan rate dependency and undefined surface oxygen state [12]. | Use a deconvolution protocol to extract intrinsic kinetics from surface oxygen effects. Perform measurements at multiple scan rates and extrapolate. |
| Poor OER activity in base | Strong scaling relationships on a single-site catalyst [11]. | Develop bimetallic catalysts (e.g., Ni-Fe) that enable dynamic dual-site cooperation to circumvent LSRs. |
| Low reproducibility of catalyst performance from lab to scale | Variations in physicochemical properties (surface area, porosity) and heat/mass transfer issues during scale-up [13]. | Implement pilot-scale testing and advanced simulation/modeling. Design for scalability from the initial R&D phase [13]. |
| Fe impurity contamination in OER tests | Unintentional incorporation of Fe from electrolytes or apparatus, altering active sites [11]. | Use high-purity KOH electrolytes and ensure all glassware is meticulously cleaned. Control Fe addition for systematic study. |
Research Reagent Solutions
The table below lists essential materials and their functions for key experiments in oxygen electrocatalysis.
| Research Reagent | Function & Application |
|---|---|
| Polycrystalline Pt Electrode / RDE | The model system for fundamental ORR studies, providing a well-defined and reproducible surface for kinetic analysis [12]. |
| Holey Graphene Nanomesh (GNM) | A support material for single-atom catalysts, offering high surface area and defect-rich sites for anchoring metal atoms [11]. |
| Ni-Fe Molecular Complex | A model bimetallic catalyst system constructed via in situ electrochemical activation, used to study dynamic site cooperation for breaking OER scaling relationships [11]. |
| High-Purity KOH Electrolyte | Essential for reliable OER testing in alkaline conditions, preventing false activity from trace metal impurities (e.g., Fe) [11]. |
| Three-Electrode Electrochemical Cell | The standard setup for half-cell measurements, consisting of a Working Electrode, Reference Electrode (e.g., RHE), and Counter Electrode (e.g., Pt wire) [14]. |
Experimental Protocols & Data
Protocol: Extracting Intrinsic ORR Kinetics on Polycrystalline Platinum
Objective: To deconvolute the intrinsic ORR kinetics from the effects of surface oxygen on Pt(pc) [12].
Methodology:
- Electrode Preparation: Use a polished glassy carbon rotating disk electrode (RDE). Clean the Pt(pc) electrode through potential cycling in a deaerated supporting electrolyte (e.g., 0.1 M HClO₄ or H₂SO₄) within a defined potential window (e.g., 0.05 - 1.4 V vs. RHE) until a stable cyclic voltammogram (CV) is obtained [14].
- Surface Characterization: Record a CV at multiple scan rates (e.g., 20-100 mV/s) in the supporting electrolyte to characterize the hydrogen underpotential deposition (H-upd) and oxygen underpotential deposition (O-upd) regions. Use techniques like scanning potential electrochemical impedance spectroscopy (SPEIS) to separate the charge associated with reversible *OH formation from irreversible *O species [12].
- ORR Activity Measurement: Saturate the electrolyte with oxygen. Record ORR polarization curves using the RDE at a rotation speed of 1600 rpm (or similar) at multiple scan rates (e.g., 1-50 mV/s). Correct all data for background and mass transport contributions to obtain the kinetic current (i_kin) [12] [14].
- Data Deconvolution: Analyze the kinetic current as a function of potential and scan rate. The intrinsic ORR current on a metallic Pt surface can be extracted by accounting for the blocking and electronic effects of surface oxygen using an equation of the form [12]:
i_ORR(E) = T_Blocking * T_Electronic * i_kin(E)
Key Quantitative Data from Protocol: Table: Intrinsic ORR Kinetic Parameters for Pt(pc) [12]
| Parameter | Value | Conditions |
|---|---|---|
| Tafel Slope | ~120 mV/decade | Extracted intrinsic value |
| Exchange Current Density (i₀) | 13 ± 4 µA/cm² | |
| Specific Activity | 7 mA/cm² | at 900 mV vs. RHE |
| O-upd Charge | ||
| - Reversible *OH | 40 ± 5 µC/cm² | |
| - Total Irreversible *O | Dominates O-upd region |
Protocol: Constructing a Ni-Fe Molecular Complex for OER
Objective: To synthesize a dynamic dual-site OER catalyst via in situ electrochemical activation and study its mechanism [11].
Methodology:
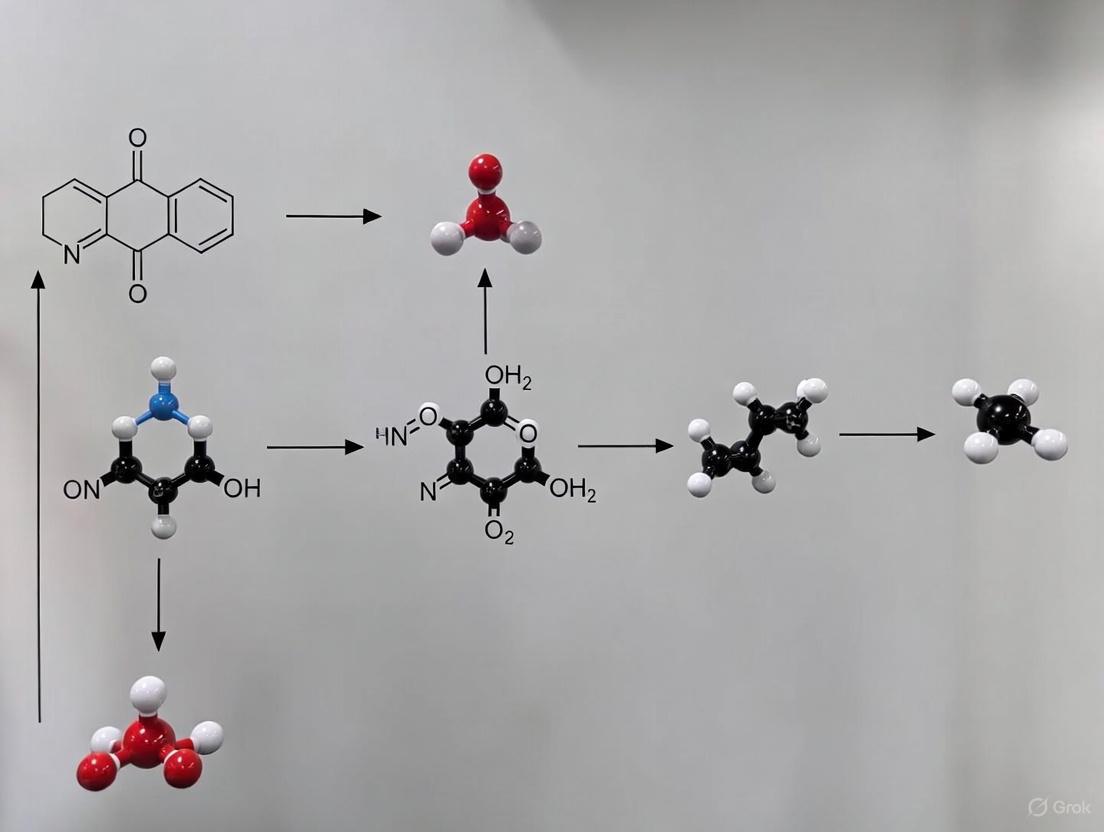
- Pre-catalyst Synthesis: a. Synthesize a Ni single-atom pre-catalyst (Ni-SAs@GNM). Create a 3D Ni(OH)₂/graphene oxide hydrogel. b. Freeze-dry the hydrogel to form an aerogel. c. Thermally anneal the aerogel at 700°C under an inert atmosphere (Ar) to form a holey graphene nanomesh (GNM) with Ni/NiO nanoparticles. d. Remove nanoparticles via acid treatment, leaving atomically dispersed Ni single atoms anchored on the GNM support [11].
- Electrochemical Activation: a. Load the Ni-SAs@GNM onto a glassy carbon working electrode. b. Use a standard three-electrode setup in a Fe-free 1 M KOH electrolyte with a deliberate addition of a low concentration (e.g., 1 ppm) of Fe ions (Fe(OH)₄⁻). c. Perform activation using cyclic voltammetry (CV) between 1.1 and 1.65 V vs. RHE. This drives Fe species to anchor onto Ni sites, forming an O-bridged Ni-Fe₂ trimer molecular complex [11].
- Characterization: a. Use operando X-ray absorption fine structure (XAFS) to verify the formation of the Ni-Fe molecular complex and track structural changes during catalysis. b. Employ density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations to understand the dynamic coordination evolution during the OER cycle [11].
Key Performance Metrics:
- The catalyst exhibits a notable intrinsic OER activity due to its ability to disrupt the conventional scaling relationships [11].
- The dynamic coordination of the Ni site with adsorbates (OH and H₂O) modulates the electronic structure of the adjacent Fe active site.
- This dual-site cooperation simultaneously lowers the free energy for O-H bond cleavage and *OOH formation, breaking the linear scaling relationship [11].
Signaling Pathways & Workflows
Diagram: Dynamic Catalyst Workflow for Breaking OER Scaling.
Diagram: OER/ORR Shared Constraint.
FAQ 1: What is the fundamental origin of the performance ceiling in single-site catalysts?
The performance ceiling in single-site catalysts arises from a fundamental constraint known as the adsorption-energy scaling relation [15] [16].
In electrocatalytic reactions like the oxygen reduction reaction (ORR) or oxygen evolution reaction (OER), the reaction proceeds through multiple intermediates (e.g., *OOH, *O, and *OH). On a catalyst with only one type of active site, the adsorption-free energies of these different intermediates are strongly linearly correlated [15]. This means the binding strength of one intermediate dictates the binding strength of all others; you cannot independently optimize the adsorption energy for each reaction step [16]. This scaling relation creates an inherent thermodynamic overpotential, as the ideal balance of energies for all intermediates cannot be achieved on a single site, leading to sluggish reaction kinetics and a performance plateau [15].
FAQ 2: How can we experimentally diagnose the limitations imposed by scaling relations?
Electrochemical Impedance Spectroscopy (EIS) is a powerful technique to probe kinetic limitations. However, traditional EIS requires equilibrium conditions, which may not reflect the catalyst's behavior during actual operation. Operando EIS, performed under real working conditions, provides more relevant insights into dynamic processes and resistances, such as charge transfer barriers linked to intermediate adsorption [17]. Furthermore, in situ synchrotron spectroscopy techniques, such as X-ray absorption fine structure (XAFS) and Fourier-transform infrared (FTIR) spectroscopy, can directly identify reaction intermediates and the electronic structure of active sites during catalysis [16]. For instance, the absence of a *OOH intermediate and the observation of a key M1–O–O–M2 intermediate can provide direct evidence for a reaction mechanism that circumvents the conventional scaling relation [16].
FAQ 3: What catalyst design strategies can break the scaling relation?
Advanced catalyst designs move beyond single-site models to break the scaling relation. Key strategies include:
| Strategy | Mechanism | Key Experimental Evidence |
|---|---|---|
| Dual-Site Mechanisms [16] | Two adjacent but distinct metal atoms (e.g., Pt and Fe) adsorb an O₂ molecule in a "side-on" configuration (M1–O–O–M2), enabling direct O–O bond breakage without forming the *OOH intermediate. | In situ SR-FTIR identified the Pt–O–O–Fe intermediate; XAFS confirmed the atomic-scale structure of N-bridged Pt = N₂ = Fe sites [16]. |
| High-Entropy Alloys (HEAs) [18] | The complex local environments created by mixing multiple metal elements (e.g., Mn, Fe, Co, Ni, Cu) creates a broad adsorption energy landscape, allowing different intermediates to be stabilized optimally on different local sites. | DFT calculations showed charge redistribution and a wide range of d-band center positions, enabling optimal adsorption for multiple NO₃RR intermediates [18]. |
| High-Density Single-Atom Catalysts [19] | Creating high loadings of stable single atoms on a support via atomic-scale self-rearrangement from metastable phases. The strong metal-support interaction can modulate electronic structure. | AC-HAADF-STEM images confirmed high-density Ir single atoms; Operando XAFS tracked charge redistribution and strong p-d-f orbital couplings during OER [19]. |
Experimental Protocol: Investigating a Dual-Site Catalyst
The following protocol outlines the synthesis and characterization of a N-bridged Pt=Fe atomic-scale bimetal assembly (ABA) designed to break the ORR scaling relation [16].
1. Synthesis of Amino-Functionalized Carbon Nanoflakes (CNF–NH₂)
- Method: Nitrate pyrene (C₁₆H₁₀) in hot HNO₃ to form trinitropyrene. Subsequently, replace the nitro groups (-NO₂) with amino groups (-NH₂) to obtain CNF–NH₂.
- Purpose: The amino groups serve as anchoring sites for metal ions.
2. Metal Precursor Chelation and Pyrolysis
- Method: Prepare a precursor solution of Fe³⁺ and [PtCl₆]²⁻ in glycol solvent. Mix this solution with the CNF–NH₂, allowing the metal cations to chelate with the -NH₂ groups during freeze-drying. Finally, pyrolyze the material in an inert atmosphere at 700°C.
- Purpose: The pyrolysis step forms the stable, nitrogen-bridged atomic-scale Pt=Fe structure.
3. Structural and Electronic Characterization
- Aberration-Corrected HAADF-STEM: Use this technique to confirm the atomic dispersion of Pt and Fe atoms and measure the intermetallic distance (typically ~2.8-2.9 Å is targeted for the dual-site mechanism).
- X-ray Absorption Spectroscopy (XAS): Perform XANES and EXAFS to determine the oxidation states of the metals and confirm the coordination environment (e.g., the absence of metal-metal bonds and presence of metal-nitrogen bonds).
4. In Situ Mechanistic Probe during ORR
- In Situ Synchrotron Radiation FTIR: Use this sensitive technique to monitor the formation (or absence) of reaction intermediates on the catalyst surface under operating conditions. The key observation is the detection of a Pt–O–O–Fe vibration and the absence of *OOH signals.
- Electrochemical Polarization Curves: Record linear sweep voltammetry (LSV) curves in an O₂-saturated electrolyte to evaluate ORR activity. Calculate the kinetic current density (Jₖ) at a specific potential (e.g., 0.95 V vs. RHE) to quantify performance gains.
5. Device Integration Test
- Method: Integrate the best-performing catalyst as an air cathode in a practical device, such as a zinc-air battery.
- Metrics: Measure the peak power density (e.g., mW cm⁻²) and cycling stability to assess real-world viability.
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Catalyst Research |
|---|---|
| Amino-Functionalized Carbon Support (e.g., CNF–NH₂) | Provides anchoring sites for metal precursors, enabling the formation of atomically dispersed metal sites after pyrolysis [16]. |
| Metal Salts (e.g., H₂PtCl₆, FeCl₃) | Serve as precursors for the active metal components. The choice of anion (e.g., Cl⁻) can influence the synthesis and final structure [16] [19]. |
| Synchrotron Radiation Beamtime | Essential for performing in situ/operando XAS and FTIR to determine atomic structure and track reaction intermediates in real-time [16]. |
| Three-Electrode Electrochemical Cell | The standard setup for fundamental electrochemical measurements. A stable reference electrode (e.g., Ag/AgCl) is critical for accurate potential control and measurement [17] [20]. |
| Ion-Exchange Membrane | Used in the assembly of advanced testing devices like anion-exchange-membrane water electrolyzers (AEMWE) to evaluate catalyst performance under industrially relevant conditions [19]. |
Visualizing the Scaling Relation and Breakthrough Strategies
The following diagrams illustrate the core problem and the catalyst design solutions.
Diagram 1: The single-site catalyst performance limitation cascade.
Diagram 2: Catalyst design strategies to overcome scaling relations.
Beyond the Limit: Methodological Strategies for Disrupting Adsorption-Energy Scaling Relations
Dynamic Structural Regulation of Active Sites via Intramolecular Proton Transfer
Linear scaling relationships (LSRs) impose fundamental limitations on multi-step catalytic reactions by creating inherent correlations between the adsorption energies of different reaction intermediates. This prevents the independent optimization of each catalytic step, thereby placing a ceiling on achievable activity and selectivity [1] [21]. Dynamic structural regulation of active sites via intramolecular proton transfer represents an emerging strategy to circumvent these limitations. By enabling real-time modulation of the electronic structure and coordination environment of catalytic centers, this approach allows simultaneous optimization of multiple reaction steps that would traditionally compete within constrained scaling relationships [1].
This technical support resource provides experimental methodologies and troubleshooting guidance for researchers investigating dynamic proton transfer processes in catalytic systems, with particular emphasis on applications in oxygen electrocatalysis and related fields where breaking scaling relationships offers transformative potential [16].
Key Concepts and Fundamental Mechanisms
Understanding Scaling Relationships in Catalysis
In conventional heterogeneous catalysis, the adsorption energies of key intermediates (e.g., *OH, *O, and *OOH in oxygen evolution reaction) are linearly correlated, creating an inherent trade-off that limits optimal catalyst design [1] [16]. For instance, strengthening *OOH adsorption typically strengthens *OH adsorption to a similar degree, making it impossible to independently optimize both interactions. This scaling relationship manifests as a fundamental bottleneck across numerous catalytic processes, including oxygen evolution reaction (OER), oxygen reduction reaction (ORR), and CO2 reduction reaction (CO2RR) [1] [22] [16].
Dynamic Structural Regulation Principle
The dynamic structural regulation strategy employs intramolecular proton transfer to trigger coordinated structural changes that simultaneously optimize the energetics of multiple catalytic steps. In the Ni-Fe system, proton transfer drives Ni-adsorbate coordination changes that modulate the electronic structure of adjacent Fe centers, thereby lowering energy barriers for both O–H bond cleavage and O–O bond formation within the same catalytic cycle [1]. This mechanism operates through:
- Real-time coordination evolution of metal centers during catalysis
- Electronic structure modulation of active sites via proton-coupled electron transfer
- Dual-site cooperation enabling simultaneous optimization of competing reaction steps
Table 1: Quantitative Effects of Dynamic Regulation in Model Catalytic Systems
| Catalytic System | Reaction | Performance Metric | Improvement vs. Conventional | Key Mechanism |
|---|---|---|---|---|
| Ni-Fe₂ molecular complex [1] | OER | Intrinsic activity | Notable enhancement | Dynamic Ni-adsorbate coordination alters Fe electronic structure |
| Pt = N₂ = Fe atomic-bridge assembly [16] | ORR | Kinetic current density @ 0.95V | ~100× vs. Pt/C | Direct O–O breakage via Pt–O–O–Fe intermediate |
| Fe-Ni@BN dual-atom catalyst [22] | CO₂RR | Selectivity to CH₄ | Enhanced | Breaks scaling between OCHO* and OCHOH* adsorption |
Experimental Protocols
Synthesis of Ni-Fe Molecular Complex Catalyst
Objective: Construct a dynamically regulated Ni-Fe molecular catalyst via in situ electrochemical activation [1].
Materials:
- Graphene oxide (GO) aqueous suspension
- Nickel vessel (for hydrothermal assembly)
- High-purity KOH electrolyte
- Fe ion source (e.g., FeCl₃·6H₂O)
- Argon gas for inert atmosphere
Procedure:
- Pre-catalyst Synthesis:
- Seal GO suspension in Ni vessel at 80°C to assemble 3D Ni(OH)₂/graphene hydrogel
- Freeze-dry resulting hydrogel to form aerogel
- Thermally anneal at 700°C under Ar atmosphere to form Ni single atoms trapped in holey graphene nanomesh (Ni-SAs@GNM)
- Apply acid treatment to remove nanoparticles, leaving atomically dispersed Ni sites
- Electrochemical Activation:
- Load Ni-SAs@GNM pre-catalyst onto glassy carbon working electrode
- Employ standard three-electrode system with purified Fe-free 1 M KOH electrolyte
- Deliberately add 1 ppm Fe ions to electrolyte
- Perform cyclic voltammetry between 1.1 and 1.65 V vs. RHE until activation complete
- Alternative: Use anodic chronopotentiometry or chronoamperometry for activation
Validation:
- Confirm atomic dispersion via aberration-corrected HAADF-STEM
- Verify Fe incorporation via synchrotron-based X-ray fluorescence (SXRF)
- Determine local structure changes via operando XAFS
Diagram 1: Ni-Fe Catalyst Synthesis Workflow
Operando XAFS Characterization
Objective: Probe dynamic structural evolution of active sites during catalysis [1].
Materials:
- Synchrotron radiation source
- Electrochemical cell with X-ray transparent windows
- Potentiostat/galvanostat
- Reference electrodes (RHE)
Procedure:
- Sample Preparation:
- Prepare thin electrode films for transmission measurements
- Ensure appropriate metal loading for optimal signal-to-noise
Data Collection:
- Acquire XANES spectra at metal K-edges under operating conditions
- Collect EXAFS data across relevant potential range
- Perform quick-scanning EXAFS for time-resolved studies
Data Analysis:
- Process data using standard demethylation procedures
- Fit EXAFS spectra to determine coordination numbers and bond distances
- Monitor changes in oxidation state via edge position shifts
Troubleshooting:
- If radiation damage occurs, reduce beam intensity or use faster scanning
- For poor signal quality, optimize sample thickness or increase integration time
Electrokinetic Analysis for Proton Transfer Studies
Objective: Quantify kinetics and identify rate-determining steps in proton-coupled electron transfer processes.
Materials:
- Potentiostat with high current resolution
- Rotating disk electrode (RDE) or rotating ring-disk electrode (RRDE)
- High-purity electrolytes with controlled proton concentrations
Procedure:
- Tafel Analysis:
- Measure steady-state polarization curves
- Plot overpotential vs. log(current density)
- Extract Tafel slopes to identify possible rate-determining steps
Reaction Order Determination:
- Measure current density at fixed overpotential while varying reactant concentration
- Plot log(current) vs. log(concentration) to determine reaction orders
Isotope Effect Studies:
- Compare kinetics in H₂O vs. D₂O-based electrolytes
- Calculate kinetic isotope effect (KIE) as kH/kD
Troubleshooting Guide: FAQs
Q1: Our catalyst shows negligible improvement in OER activity after Fe incorporation. What could be wrong?
A1: Several factors could explain limited activity enhancement:
- Insufficient Fe anchoring: Ensure your Ni pre-catalyst has adequate anchoring sites. Characterize pre-catalyst with HAADF-STEM to confirm atomic dispersion [1].
- Improper electrochemical activation: Extend activation cycles or try potentiostatic activation at intermediate potentials (1.3-1.4 V vs. RHE) [1].
- Fe contamination in electrolyte: Use ultra-high purity KOH and validate Fe-free conditions before deliberate Fe addition.
Q2: Operando XAFS shows no evidence of dynamic structural changes during catalysis. What are potential causes?
A2: The absence of observable dynamics may stem from:
- Insufficient time resolution: Dynamic coordination changes can be transient. Implement quick-scanning EXAFS or dispersive XAFS for better time resolution [1].
- Inappropriate potential window: Ensure you're scanning potentials relevant to the catalytic turnover.
- Limited proton mobility: Incorporate proton donors/acceptors in proximity to active sites to facilitate intramolecular proton transfer [1] [23].
Q3: The catalyst exhibits excellent initial activity but rapid degradation. How can stability be improved?
A3: Stability issues in dynamically regulated catalysts often originate from:
- Metal dissolution: Implement strong metal-support interactions through appropriate coordination environments (e.g., N-bridging) [16].
- Structural collapse: Ensure support stability by using corrosion-resistant materials like heteroatom-doped carbons [24].
- Active site aggregation: Introduce spatial confinement through molecular scaffolds or coordination templates [1] [16].
Q4: How can we distinguish intramolecular proton transfer from intermolecular pathways?
A4: Several experimental approaches can differentiate these mechanisms:
- Isotope effect studies: Intramolecular proton transfer typically exhibits smaller KIE (2-4) versus intermolecular pathways (>7) [23].
- Solvent dependence: Intramolecular processes show minimal solvent dependence compared to intermolecular pathways [23].
- Structural constraints: Systematically vary distance between proton donor and acceptor sites; intramolecular transfer requires precise spatial arrangement [1] [23].
Q5: Our theoretical calculations suggest broken scaling relationships, but experimental performance remains limited. Why?
A5: This discrepancy may arise from:
- Non-ideal reaction pathways: The catalyst may follow alternative mechanisms that bypass the theoretically optimized pathway.
- Mass transport limitations: Ensure measurements are conducted in kinetically controlled regime using RDE at appropriate rotation rates.
- Incomplete active site formation: Characterize actual active sites under working conditions; theoretical models often assume ideal structures that may not form experimentally [1].
Research Reagent Solutions
Table 2: Essential Research Reagents for Dynamic Proton Transfer Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Single-Atom Pre-catalysts | Ni-SAs@GNM, Fe-SAs@GNM [1] | Foundation for constructing molecular complexes | Atomic dispersion critical; characterize with HAADF-STEM |
| Metal Precursors | H₂PtCl₆, FeCl₃·6H₂O, Ni salts [16] | Introduce active metal centers | Purity essential to avoid unintended doping |
| Molecular Bridges | Nitrogen-containing ligands [16] | Create defined atomic spacing for dual-site mechanisms | Coordination strength affects stability under potential cycling |
| High-Purity Electrolytes | Fe-free KOH, purified H₂SO₄ [1] | Provide reaction environment and protons | Trace metal contaminants can poison active sites |
| Isotope-labeled Solvents | D₂O, H₂¹⁸O [23] | Mechanistic studies through KIE and oxygen tracking | Exclusion of atmospheric contaminants critical |
| Support Materials | Graphene nanomesh, N-doped carbon [1] [16] | Anchor single atoms and facilitate electron transfer | Defect engineering enhances metal-support interactions |
Advanced Applications and Mechanism Elucidation
Dual-Site Mechanism in Oxygen Reduction
The N-bridged Pt = N₂ = Fe atomic-scale assembly exemplifies how dynamic regulation enables alternative reaction mechanisms that circumvent conventional scaling relationships. This system promotes direct O–O bond breakage without forming *OOH intermediates, following a dual-site mechanism characterized by a key Pt–O–O–Fe transition state [16]. The interatomic distance between Pt and Fe (∼2.8-2.9 Å) is critical for enabling this pathway, which demonstrates nearly two orders of magnitude enhancement in kinetic current density compared to conventional Pt/C catalysts [16].
Diagram 2: Dual-Site vs Single-Site ORR Mechanisms
Computational Modeling Guidance
DFT Protocol for Proton Transfer Systems:
- Model Construction:
- Build cluster models with explicit second-sphere coordination
- Include sufficient support environment to capture metal-support interactions
- Model solvation effects using implicit or explicit solvent models
Reaction Pathway Mapping:
- Identify possible proton transfer pathways and associated energy barriers
- Calculate scaling relationships between key intermediates
- Model dynamic structural changes through constrained optimization or ab initio MD
Electronic Structure Analysis:
- Compute projected density of states to identify orbital interactions
- Calculate Bader charges to track electron redistribution during proton transfer
- Analyze charge density differences to visualize bonding changes
Machine Learning Integration:
- Use descriptor-based models to rapidly screen candidate systems
- Implement neural network potentials for accurate dynamics simulations
- Apply symbolic regression to discover new scaling relationships
The strategic application of dynamic structural regulation via intramolecular proton transfer represents a paradigm shift in catalyst design that directly addresses the fundamental limitations imposed by linear scaling relationships. The experimental protocols, troubleshooting guidelines, and mechanistic insights provided in this technical resource establish a foundation for systematic investigation of these complex dynamic systems. As research in this field advances, the integration of sophisticated operando characterization with computational modeling will continue to unravel the intricate interplay between proton transfer, structural dynamics, and catalytic function, enabling the rational design of next-generation catalysts with transformative performance capabilities across energy conversion and chemical transformation technologies.
FAQs: Understanding the Mechanism and Its Challenges
Q1: What is the primary kinetic advantage of bypassing the *OOH intermediate in the Oxygen Reduction Reaction (ORR)?
A1: Bypassing the *OOH intermediate via a dissociative mechanism avoids the rate-limiting step of its formation, which traditionally requires overcoming a high Gibbs free energy barrier (ΔG) [25]. This mechanism facilitates direct O-O bond cleavage, enabling rapid ORR kinetics. In practice, catalysts employing this pathway, such as those with abundant sp3-hybridized carbon defects, have demonstrated excellent performance with onset potentials of 1.02 V and half-wave potentials of 0.90 V [25].
Q2: How do dual-site catalysts fundamentally differ from single-atom catalysts in managing reaction intermediates?
A2: Single-atom catalysts feature uniform active sites, which can limit performance in complex reactions involving multiple intermediates because they cannot optimally adjust the adsorption of every intermediate simultaneously [6]. In contrast, dual-site catalysts, or Integrative Catalytic Pairs (ICPs), feature spatially adjacent, electronically coupled active sites that function cooperatively [6]. This allows for functional differentiation within the catalytic ensemble, enabling the system to stabilize different transition states or intermediates at different sites, thereby circumventing the linear scaling relationships (LSRs) that constrain single-site catalysts [6] [11].
Q3: What are the key characterization techniques for verifying a direct O-O cleavage pathway and the dynamic nature of active sites?
A3: A combination of advanced techniques is required:
- Operando X-ray Absorption Fine Structure (XAFS): Essential for probing the local coordination environment of metal active sites during electrochemical operation. This technique can verify dynamic structural changes, such as the evolution of a Ni-Fe2 trimer structure during OER [11].
- Density Functional Theory (DFT) Calculations: Used to map reaction pathways, calculate the free energy (
ΔG) of reaction steps, and identify the critical role of orbital interactions. For instance, DFT can show how sp3 carbon provides electrons to oxygen's π* and σ* anti-bonding orbitals, facilitating O2 adsorption and OH desorption [25]. - Electrokinetic Studies: Analyzing parameters like Tafel slopes can help distinguish between different reaction mechanisms and rate-determining steps [11].
Troubleshooting Guides for Experimental Research
Common Experimental Challenges and Solutions
When working with advanced catalytic systems designed for direct O-O cleavage, researchers may encounter several challenges. The table below outlines common issues, their potential causes, and recommended corrective actions.
| Problem | Potential Cause | Solution |
|---|---|---|
| Low Catalytic Activity | • Insufficient density of active sites.• Incorrect electronic structure of metal centers.• Catalyst surface area is too low. | • Optimize synthesis to increase sp3 carbon content or dual-site density [25].• Fine-tune the coordination environment of metal centers via precursor selection [11].• Use template-assisted strategies to achieve high specific surface area (e.g., 1120 m²/g) [25]. |
| Poor Stability During Electrolysis | • Degradation or agglomeration of active sites.• Structural collapse of the support material.• Unfavorable reaction intermediates causing poisoning. | • Implement a stabilizing matrix (e.g., holey graphene nanomesh) [11].• Perform operando characterization to identify degradation pathways [11]. |
| Inability to Detect Key Intermediates | • Intermediates are too short-lived.• Low concentration of intermediates on the surface.• Inappropriate characterization technique. | • Utilize computational methods (DFT) to predict intermediate stability and binding energies [25].• Employ surface-sensitive in situ techniques (ATR-SEIRAS). |
| Failure to Break Scaling Relationships | • Active sites are too uniform (single-site limitation).• Lack of dynamic cooperation between adjacent sites.• Inter-site distance is not optimal for bridge-model bonding. | • Design catalysts with integrative catalytic pairs (ICPs) for functional differentiation [6].• Aim for systems that exhibit dynamic structural regulation under reaction conditions [11]. |
Step-by-Step Experimental Protocol: Synthesis of N-doped Defective Carbon with sp3-Hybridized Defects
This protocol is adapted from the synthesis of a metal-free carbon electrocatalyst with high sp3 content, which demonstrated high ORR activity via a dissociative mechanism [25].
Objective: To fabricate graphene-like, N-doped defective carbon with abundant pentagonal sp3 carbon structures via high-temperature pyrolysis of silk using a vaporized-salt template.
Materials:
- Silk (source of carbon and nitrogen)
- Deionized (DI) water
- KCl (2 M solution)
- LiCl (2 M solution)
- Tube furnace with N2 atmosphere
- Aqueous HCl (1 M)
Procedure:
- Silk Pre-treatment: Immerse the silk in DI water for 24 hours. Afterwards, dry it at 60°C for 6 hours.
- Salt Impregnation: Cut the washed silk into pieces (2 cm x 2 cm). Immerse them in a 60 mL mixed solution containing 2 M KCl and 2 M LiCl for 24 hours.
- Drying: Transfer the mixture to a culture dish and dry at 60°C.
- Pyrolysis: Place the dried sample in a tube furnace and anneal at 900°C for 2 hours under a continuous N2 atmosphere to form graphene-like carbon (GC).
- Purification: To remove potential metal impurities, treat the obtained sample with 1 M aqueous HCl solution, followed by thorough washing with DI water and drying.
Key Characterization:
- The successful incorporation of sp3-hybridized carbon (content can reach 43.7%) can be confirmed by X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy [25].
- The specific surface area (BET method) of the final material can be as high as 1120.83 m²/g [25].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key materials and reagents used in the synthesis and testing of advanced catalysts for direct O-O cleavage, as featured in the cited research.
| Research Reagent / Material | Function / Role in the Experiment |
|---|---|
| Silk (as a biomass precursor) | Serves as a source of both carbon and nitrogen for creating N-doped carbon frameworks with inherent heteroatoms and molecular structures that can form topological defects [25]. |
| KCl / LiCl (mixed salt template) | Acts as a vaporized-salt template during high-temperature pyrolysis. This process helps create ultra-thin, high-surface-area carbon structures with abundant defects [25]. |
| Graphene Oxide (GO) Suspension | Used as a foundational support material for constructing single-atom pre-catalysts. It can be formed into a 3D hydrogel and subsequently processed into a holey graphene nanomesh [11]. |
| Fe ions (e.g., from Fe salts) | Deliberately added in ppm quantities to an electrolyte to enable the in situ electrochemical construction of bimetallic active sites, such as the O-bridged Ni-Fe2 trimer complex [11]. |
| Purified KOH Electrolyte | Used for electrochemical activation and testing under alkaline conditions. High purity is critical to avoid unintended contamination by trace metals that could influence catalyst formation [11]. |
Catalytic Pathway Visualization
Diagram 1: Dissociative ORR pathway bypassing *OOH intermediate.
Diagram 2: Dynamic cooperation mechanism in Ni-Fe catalyst for OER.
Engineering Multifunctional Surfaces and Interfacial Sites for Intermediate Stabilization
This technical support center provides troubleshooting guides and FAQs for researchers developing advanced catalysts to overcome linear scaling relationships (LSRs) in multi-step reactions.
Frequently Asked Questions (FAQs)
Q1: Our catalyst synthesis consistently yields inconsistent Ni-Fe distributions. What could be the cause? Inconsistent atomic distribution in bimetallic catalysts like Ni-Fe complexes often stems from non-uniform precursor deposition or inadequate control during the electrochemical activation step. Ensure your graphene oxide support has uniform defect sites for metal anchoring and control the Fe ion concentration precisely in the ppm range during electrochemical activation. Using a purified KOH electrolyte is essential to prevent unintended metal contamination that competes with active site formation [11].
Q2: We observe a gradual decline in catalyst conversion efficiency during oxygen evolution reaction (OER). What troubleshooting steps should we follow? A gradual decline in activity often indicates catalyst deactivation. Systematically check these parameters [26]:
- Thermal degradation: Verify operating temperatures haven't exceeded design limits causing sintering
- Chemical poisoning: Analyze feed composition for contaminants (sulfur compounds commonly poison metal catalysts)
- Structural changes: Perform post-reaction characterization to check for active site agglomeration or carbon buildup
- Operando analysis: Implement techniques like X-ray absorption fine structure (XAFS) to monitor active site evolution under reaction conditions [11]
Q3: What is the significance of the water/chloride molar ratio in catalytic reforming, and how is it controlled? Maintaining proper water/chloride balance (typically 15-25 molar ratio) is crucial for balancing the acidic and metal functions of reforming catalysts. This is controlled through:
- Online monitoring systems for water and chloride concentrations in recycle gas
- Sample facilities for regular compositional analysis
- Following licensor-specific equilibrium curves for chloride management
- Adjusting water injection rates based on catalyst chloride content and operating temperature [27]
Q4: How can we verify if channeling is occurring in our fixed-bed catalytic reactor? Channeling in fixed-bed reactors can be confirmed through these diagnostic methods:
- Measure temperature variations across the reactor bed - variations exceeding 6-10°C indicate flow maldistribution
- Monitor differential pressure trends - unexpected DP changes often signal channeling
- Check for difficulty meeting product specifications despite normal operating parameters
- Implement a well-designed pattern of radial bed thermocouples for accurate monitoring [26]
Troubleshooting Guides
Catalyst Synthesis and Characterization Issues
| Symptom | Possible Causes | Diagnostic Methods | Solutions |
|---|---|---|---|
| Low catalytic activity | Incorrect metal coordination, Sintering of active sites, Poisoning by feed impurities [26] | XAFS, XPS, ICP-OES [11] | Optimize electrochemical activation, Use purified electrolytes, Control feed quality [11] |
| Poor stability | Phase transformations, Carbon buildup, Active species volatilization [26] | In-situ XAFS, TEM, TPO [11] | Modify support interactions, Introduce protective layers, Control reaction severity [28] |
| Unselective product distribution | Unbalanced acid/metal sites, Incorrect intermediate stabilization [26] | Kinetic analysis, Isotope labeling, DFT calculations [11] | Tune water/chloride ratio, Engineer dual active sites, Optimize operating conditions [27] [11] |
Reactor Operation and Performance Issues
| Symptom | Possible Causes | Diagnostic Methods | Solutions |
|---|---|---|---|
| Temperature runaway | Loss of quench gas, Hot spots from flow maldistribution, Feed composition changes [26] | Radial temperature profiling, Feed analysis, Pressure monitoring [26] | Install better flow distributors, Implement feed quality control, Ensure adequate cooling capacity [26] |
| Rapid pressure drop increase | Catalyst fines from poor loading, Feed precursors for polymerization, Channeling [26] | DP monitoring, Catalyst sampling, Radioactive tracing | Improve catalyst loading procedures, Implement feed pre-treatment, Add in-line filters [26] |
| Unexpected selectivity changes | Feed contaminants, Altered water/chloride balance, Catalyst poisoning [26] [27] | Recycle gas analysis, Catalyst characterization, Feed impurity testing | Install guard beds, Adjust water injection rates, Implement stricter feed specifications [27] |
Experimental Protocols
Protocol 1: Synthesis of Ni-Fe Molecular Complex Catalyst via Electrochemical Activation
Principle: Construct dynamic dual-site catalysts through in situ electrochemical conversion of single-atom precursors to achieve intermediate stabilization beyond LSRs [11].
Materials:
- Graphene oxide (GO) aqueous suspension
- High-purity Ni vessel
- Argon atmosphere furnace
- Purified Fe-free 1 M KOH electrolyte
- Fe standard solution for precise ppm-level addition
- Standard three-electrode electrochemical cell
Procedure:
- Pre-catalyst Preparation:
- Seal GO suspension in Ni vessel at 80°C for 24 hours to form 3D Ni(OH)₂/graphene hydrogel
- Freeze-dry the resulting hydrogel to preserve structure
- Thermally anneal at 700°C under Ar atmosphere to form Ni single atoms on holey graphene nanomesh (Ni-SAs@GNM)
- Apply acid treatment to remove nanoparticles, leaving only atomic Ni dispersions
Electrochemical Activation:
- Load Ni-SAs@GNM onto glassy carbon working electrode
- Use purified 1 M KOH electrolyte with deliberate addition of 1 ppm Fe ions
- Perform cyclic voltammetry between 1.1 and 1.65 V vs. RHE until stable OER performance achieved
- Alternative activation methods: anodic chronopotentiometry or chronoamperometry
Characterization:
- Confirm atomic dispersion via aberration-corrected HAADF-STEM
- Determine oxidation states through XPS and L₂,₃-edge XAS
- Verify local structure via operando XAFS measurements
- Elemental distribution analysis through SXRF spectroscopy and EDS mapping [11]
Protocol 2: Operando XAFS Analysis for Dynamic Active Site Monitoring
Principle: Track the coordination evolution of active sites during catalysis to understand dynamic structural changes that enable LSR circumvention [11].
Materials:
- Synchrotron radiation facility with operando electrochemical cell
- Custom-designed Teflon cell with X-ray transparent windows
- Potentiostat for potential control during measurements
- Reference electrodes (RHE) calibrated immediately before experiments
Procedure:
- Cell Assembly:
- Prepare electrode with catalyst loading optimized for X-ray absorption
- Integrate working, counter, and reference electrodes in operando cell
- Ensure precise alignment for X-ray path through electrode interface
Data Collection:
- Acquire Ni K-edge and Fe K-edge spectra under open circuit conditions
- Collect data at progressively increasing applied potentials covering OER region
- Perform quick-scanning EXAFS to capture transient intermediates
- Record simultaneous electrochemical data (current, potential)
Data Analysis:
- Process data using standard procedures (background subtraction, normalization)
- Perform linear combination analysis to identify species evolution
- Extract structural parameters (coordination numbers, bond distances) through EXAFS fitting
- Correlate structural changes with electrochemical performance [11]
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| Graphene Oxide Support | Provides high-surface-area with defined anchoring sites | Ensure uniform holey structure with 20-60 nm pores for optimal metal dispersion [11] |
| Ni-Fe Molecular Complex | Dynamic dual-site OER catalyst | Synthesized via electrochemical activation; enables intramolecular proton transfer [11] |
| Operando XAFS Setup | Monitors atomic-scale structural changes during operation | Requires synchrotron source; provides real-time coordination environment data [11] |
| Purified KOH Electrolyte | Prevents contamination during electrochemical activation | Essential for controlled Fe incorporation at ppm levels [11] |
| Single-Atom Pre-catalysts | Precursors for complex active sites | Ni-SAs@GNM provides defined starting point for electrochemical transformation [11] |
Performance Metrics for Advanced Catalysts
| Catalyst System | OER Activity | Stability | Key Innovation | LSR Mitigation Strategy |
|---|---|---|---|---|
| Ni-Fe Molecular Complex [11] | Notable intrinsic OER activity | Maintained under operation | Dynamic dual-site cooperation | Intramolecular proton transfer alters electronic structure |
| Cu₃N (001) Surface [29] | Better HER than Pt-based catalysts | Not specified | Multifunctional capability | Unique N and Cu atom coordination |
| Integrative Catalytic Pairs [6] | Enhanced multi-step reaction efficiency | Improved selectivity | Spatially adjacent dual sites | Functional differentiation within ensemble |
| LDH-Based Composites [28] | Enhanced photocatalytic H₂ production | Tunable stability | Surface and interface engineering | Band alignment tailoring and defect creation |
Diagnostic Diagrams
Catalyst Troubleshooting Diagnostic Flow
Breaking Scaling Relationships via Surface Engineering
Utilizing Confinement Effects and Proton Acceptors to Alter Adsorption Geometries
This technical support center provides troubleshooting guidance for researchers working to overcome catalytic scaling relationships by manipulating adsorption geometries.
Frequently Asked Questions & Troubleshooting
Q1: My catalyst shows unwanted adsorption geometry, leading to poor selectivity. How can I influence this?
- Problem: The reactant molecules adsorb onto the active site in a geometry that leads to an undesired reaction pathway or product.
- Solution & Protocol: Implement spatial confinement using 1D nanochannels.
- Select a Confining Support: Choose a support material with well-defined, tunable nanochannels, such as ultra-thin C4N3 nanotubes [30] or Carbon Nanotubes (CNTs) [31].
- Functionalize the Nanochannel: Anchor single-atom catalytic sites (e.g., Transition Metal (TM) atoms like Mn, Cr, or Mo) to the inner wall of the nanochannels to create a confined reaction environment [30].
- Mechanism: The nanoscale walls of the channel physically constrain how reactant molecules can approach and orient themselves on the active site. This can force a switch from an "end-on" to a more favorable "side-on" adsorption geometry for molecules like N₂, which is crucial for activating strong covalent bonds [30].
Q2: My highly reactive catalyst deactivates rapidly during operation. How can confinement improve stability?
- Problem: The catalyst, especially those used in aggressive reactions like Advanced Oxidation Processes (AOPs), loses activity due to leaching of active species or over-oxidation.
- Solution & Protocol: Fabricate a catalytic membrane with angstrom-scale confinement.
- Synthesis: Intercalate catalyst nanoparticles (e.g., Iron Oxyfluoride, FeOF) between layers of Graphene Oxide (GO) to create a layered membrane with sub-1 nm channels [32].
- Operation: Operate in a flow-through mode where the reaction mixture is forced through the confined channels [32].
- Mechanism: The confined space acts as a nano-reactor that restricts the mobility and leaching of active species (e.g., fluoride ions in FeOF). It also protects the active sites from deactivating interactions with large, interfering molecules in the solution, thereby significantly extending catalyst lifetime [32].
Q3: The reaction rate in my confined catalyst is lower than expected. What could be the issue?
- Problem: While confinement improves selectivity, it can sometimes lead to mass transfer limitations that slow down the overall reaction rate.
- Solution & Protocol: Optimize the nanochannel diameter and surface properties.
- Diameter Tuning: There is a volcano-type relationship between channel diameter and activity. If the channel is too small (<1 nm), it can weaken reactant-catalyst binding; if too large (>100 nm), confinement effects are lost. Perform simulations (e.g., DFT, molecular dynamics) to find the optimal diameter for your specific reaction [31].
- Surface Engineering: Modify the inner surface of the nanochannels to enhance the transport of key species. Introducing specific functional groups can create a "quantum-confined superfluid" state, leading to ultrafast, directional flow of reactants [31].
Q4: How can I control proton transfer to steer a Proton-Coupled Electron Transfer (PCET) reaction?
- Problem: The reaction mechanism is highly sensitive to proton availability, leading to unwanted intermediates or pathways.
- Solution & Protocol: Carefully select the proton donor/acceptor species in your electrolyte.
- Parameter Selection: The concentration, pKa, and steric structure of proton donors/acceptors (e.g., solvents, buffers) can profoundly alter the PCET pathway [33].
- Mechanism Control: Using a proton donor with a suitable pKa can promote a more energetically favorable concerted proton-electron transfer (CPET) pathway, avoiding high-energy intermediates that occur in stepwise (PTET or ETPT) mechanisms [33].
- Construction of Proton Channels: Design catalysts with built-in proton transfer channels, mimicking enzymatic active sites. This can involve organizing water networks or functional groups to shuttle protons directly to the reaction center [33].
Experimental Protocols
Protocol 1: Designing Transition Metal Endohedral Nanotubes for N₂ Adsorption
This protocol outlines the computational design and screening of single-atom catalysts confined in ultra-thin nanotubes for altering dinitrogen adsorption geometry, based on methodologies in [30].
1. Model Construction:
- Build an atomic model of an ultra-thin armchair (3,3) C4N3 nanotube (CNNT).
- Embed a single transition metal (TM) atom (e.g., Mn, Fe, Cr, Mo, W) into the intrinsic pore on the nanotube's inner surface, forming an MN₃-type active site.
2. Computational Settings (DFT):
- Use the Vienna ab initio Simulation Package (VASP).
- Apply the Perdew-Burke-Ernzerhof (PBE) functional for electron exchange-correlation.
- Set a plane-wave basis set cutoff energy to 400 eV.
- Use a 1x1x5 k-point mesh for Brillouin zone sampling.
- Include Grimme's DFT-D3 method for van der Waals corrections.
- Optimize all atomic coordinates until the force on each atom is less than 0.02 eV/Å.
3. Screening Procedure:
- Step 1 - Stability Check: Calculate the binding energy (Ebind) of the TM atom. Discard configurations with positive Ebind (unstable).
- Step 2 - N₂ Adsorption Assessment: Calculate the adsorption energy (Ead) of an N₂ molecule on the TM site. Evaluate the stable adsorption geometry (end-on vs. side-on).
- Step 3 - Activity Proxy: Calculate the free energy change (ΔG) for the first hydrogenation step from *N₂ to *N₂H. This step is often a potential-determining step.
- Step 4 - HER Competition: Calculate the free energy of hydrogen adsorption (ΔG*H) to evaluate the catalyst's tendency to perform the competing Hydrogen Evolution Reaction (HER).
4. Expected Outcome: This screening workflow will identify promising catalyst candidates (e.g., Mn-CNNT, which favors a side-on N₂ adsorption geometry) for further experimental investigation [30].
Protocol 2: Fabricating an Angstrom-Confined Catalytic Membrane
This protocol describes the synthesis of a spatially confined catalytic membrane for enhancing catalyst stability in water treatment applications, based on [32].
1. Catalyst Synthesis (Iron Oxyfluoride - FeOF):
- Place FeF₃·3H₂O in a methanol medium within an autoclave.
- Heat at 220 °C for 24 hours.
- Recover the synthesized FeOF powder, and confirm its crystalline structure and layered morphology via X-ray Diffraction (XRD) and Scanning/Transmission Electron Microscopy (SEM/TEM).
2. Membrane Fabrication:
- Prepare a suspension of single-layer Graphene Oxide (GO) sheets in water.
- Disperse the synthesized FeOF catalyst into the GO suspension.
- Use vacuum-assisted filtration to assemble the mixture into a layered membrane structure. The GO sheets stack, creating aligned nanochannels (width < 1 nm) with the FeOF catalysts confined between them.
3. Performance Evaluation:
- Setup: Operate the membrane in a flow-through filtration cell.
- Reaction: Pass a solution containing the target pollutant (e.g., neonicotinoid insecticides) and an oxidant (e.g., H₂O₂) through the membrane.
- Analysis:
- Monitor pollutant removal efficiency over time using techniques like High-Performance Liquid Chromatography (HPLC).
- Use Electron Paramagnetic Resonance (EPR) spectroscopy with DMPO as a spin trap to quantify the generation of reactive oxygen species (e.g., •OH).
- Analyze the membrane post-operation for catalyst leaching (using ICP-OES for Fe and Ion Chromatography for F) and structural integrity (via XRD, XPS).
The Scientist's Toolkit: Key Research Reagents & Materials
Table 1: Essential materials and their functions in experiments involving confinement and proton transfer.
| Reagent/Material | Function/Explanation | Key Reference |
|---|---|---|
| C4N3 Nanotubes | An ultra-thin, semi-metallic 1D support. Its periodic pores are ideal for anchoring single metal atoms, creating a confined nano-reactor. | [30] |
| Graphene Oxide (GO) | A 2D material used to create layered membranes with tunable angstrom-scale channels for spatial confinement of catalysts. | [32] |
| Transition Metal Atoms | Single atoms (e.g., Mn, Fe, W) serve as the primary active sites for reactant adsorption and activation within the confined space. | [30] |
| Iron Oxyfluoride (FeOF) | A highly efficient, layered catalyst for activating peroxides. Its stability is greatly enhanced by spatial confinement. | [32] |
| Proton Donors/Acceptors | Molecules (e.g., specific solvents, buffers) that provide or accept protons during PCET reactions, steering the reaction mechanism and kinetics. | [33] |
Experimental Workflow and Data
Confinement Effect Experimental Workflow
Quantitative Data on Confinement Effects
Table 2: Experimentally observed performance enhancements due to confinement effects.
| System Description | Key Performance Metric | Result with Confinement | Result without Confinement / Bulk | Reference |
|---|---|---|---|---|
| FeOF confined in GO membrane (Advanced Oxidation) | Catalyst Stability (Pollutant Removal) | Maintained >90% removal over 2 weeks | ~75% activity loss after 1-2 cycles (powder form) | [32] |
| FeOF confined in GO membrane | Radical Generation (EPR signal intensity) | Sustained high •OH generation | 70.7% decrease in 2nd run (powder form) | [32] |
| Reaction in Carbon Nanotubes | Selectivity (para-bromination) | 97% selectivity | 68% selectivity | [31] |
| TM atoms in C4N3 Nanotubes | N₂ Adsorption Geometry | Favors selective side-on configuration (e.g., on Mn site) | Often less selective end-on configuration | [30] |
Designing Atomic-Scale Bimetal Assemblies with Tailored Intermetallic Distances
A fundamental challenge in modern electrocatalysis is the presence of linear scaling relationships (LSRs) between the adsorption energies of reactive intermediates. These relationships create an inherent thermodynamic limitation for multi-step reactions, particularly the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), which are crucial for sustainable energy technologies like fuel cells and metal-air batteries [34] [15] [5]. The binding energies of intermediates such as *OOH, *O, and *OH on conventional single-site catalysts are strongly correlated, making it impossible to independently optimize the adsorption strength for each intermediate to achieve maximal catalytic activity [15] [5].
This technical support document explores how designing atomic-scale bimetal assemblies (ABAs) with precisely controlled intermetallic distances presents a groundbreaking strategy to circumvent these scaling relationships. By enabling a dual-site reaction mechanism that bypasses certain intermediates entirely, this approach enables the development of catalysts with dramatically enhanced kinetics and selectivity [34] [1].
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary experimental strategies for creating atomic-scale bimetal assemblies?
- The most common approach involves a pyrolysis-based synthesis on functionalized carbon supports. For instance, one documented method uses amino-functionalized carbon nanoflakes (CNF‑NH₂) to anchor metal precursors, followed by controlled thermal treatment to form stable N-bridged structures like Pt = N₂ = Fe [34]. Alternatively, in situ electrochemical activation of pre-catalysts in electrolyte solutions containing specific metal ions can construct dynamic bimetallic sites, such as the conversion of Ni single atoms to O-bridged Ni-Fe₂ trimers [1].
FAQ 2: What techniques provide atomic-scale confirmation of successful assembly formation?
- Cs-corrected STEM-HAADF imaging allows direct visualization of individual metal atoms and measurement of interatomic distances (e.g., confirming Pt-Fe distances of 2.83–2.91 Å) [34].
- Synchrotron-based techniques including X-ray absorption fine structure (XAFS) spectroscopy, particularly in operando mode, provide evidence for hypothesized structures and their evolution under reaction conditions by analyzing oxidation states and local coordination environments [34] [1].
FAQ 3: Our bimetal catalyst isn't achieving the expected performance. Where should we focus our troubleshooting?
- First, verify the intermetallic distance falls within the optimal 2.8–3.0 Å range using STEM and XAFS, as distances that are too long or too short favor single-site mechanisms or inefficient pathways [34].
- Second, use in situ characterization like SR-FTIR to confirm the reaction mechanism; the absence of a *OOH intermediate signal and detection of a M1–O–O–M2 species indicates successful dual-site mechanism operation [34].
- Third, ensure sufficient density of dual sites; statistical analysis of STEM images should show a high percentage (e.g., ~73% reported in one study) of sites comprising both metals rather than isolated single atoms [34].
FAQ 4: How does the dual-site mechanism fundamentally break the scaling relationship?
- In conventional ORR on single-site catalysts, the reaction proceeds through a defined sequence of intermediates (O₂ → *OOH → *O → *OH) where *OOH and *OH binding energies are linearly scaled [34] [15]. The dual-site mechanism enables direct O–O bond breakage upon adsorption, forming a key M1–O–O–M2 intermediate and completely bypassing the *OOH formation step. This bypasses the thermodynamic limitation imposed by the *OOH-OH scaling relationship [34].
Troubleshooting Common Experimental Challenges
Problem 1: Formation of Monometallic Sites Instead of Desired Bimetal Assemblies
| Potential Cause | Diagnostic Methods | Recommended Solution |
|---|---|---|
| Insufficient functionalization of carbon support | FTIR to confirm presence of amine groups (-NH₂) [34] | Optimize nitration and amination steps during CNF-NH₂ synthesis; verify metal-chelation capability [34] |
| Improper metal precursor ratio or affinity | ICP-OES to measure final metal content vs. target [34] | Use precursor solutions promoting affinity between hetero-electric metals (e.g., Fe³⁺ and [PtCl₆]²⁻ in glycol solvent) [34] |
| Suboptimal pyrolysis conditions | XAFS to check for unwanted metallic nanoparticles or oxides [34] | Systematically vary pyrolysis temperature and atmosphere; characterize with XRD and HRTEM to confirm atomic dispersion [34] |
Problem 2: Poor Catalytic Activity Despite Successful Assembly Formation
| Potential Cause | Diagnostic Methods | Recommended Solution |
|---|---|---|
| Suboptimal intermetallic distance | STEM-HAADF to measure atomic spacing; XAFS for local structure [34] | Tune bridge ligands (e.g., N-bridging vs. O-bridging) during synthesis to achieve ideal ~2.8-2.9 Å distance [34] |
| Dynamic structural changes under operating conditions | Operando XAFS to monitor state during electrochemical testing [1] | Employ supports that stabilize structures during reaction; explore different metal pairs with higher inherent stability |
| Low density of active sites | Statistical analysis of STEM images; electrochemical active site quantification [34] | Increase density of functional groups on support; optimize metal loading during precursor deposition |
Problem 3: Inconsistent Results Between Experimental Batches
| Potential Cause | Diagnostic Methods | Recommended Solution |
|---|---|---|
| Variations in carbon support morphology | SEM/TEM to compare layered morphology; BET surface area analysis [34] | Standardize synthesis of carbon nanoflakes; implement rigorous quality control for precursor materials |
| Trace metal contamination | ICP-MS/OES of electrolytes and reagents; SXRF mapping [1] | Use high-purity reagents (e.g., Fe-free KOH); employ controlled environments to prevent contamination during synthesis |
| Inconsistent thermal treatment | Calibration of pyrolysis furnace; XRD to detect crystalline impurities [34] | Implement precise temperature ramping protocols; use uniform reactor geometry across batches |
Experimental Protocols & Data Interpretation
Synthesis Protocol: N-Bridged Pt = N₂ = Fe Atomic Bimetal Assembly
This protocol creates atomic-scale bimetal assemblies with controlled intermetallic distances for enhanced oxygen reduction reaction [34].
Materials:
- Pyrene (C₁₆H₁₀) as carbon feedstock
- Nitric acid (HNO₃) for nitration
- Reducing agents for amino functionalization
- Metal precursors: H₂PtCl₆ and FeCl₃·6H₂O
- Glycol solvent
- Inert gas (Ar or N₂) for pyrolysis
Step-by-Step Procedure:
- Nitration of Pyrene: Treat pyrene with hot HNO₃ to form trinitropyrene with high activation state.
- Amino Functionalization: Replace nitro groups (-NO₂) with amino groups (-NH₂) to create amino-functionalized carbon nanoflakes (CNF‑NH₂). Confirm complete replacement by FTIR [34].
- Metal Precursor Preparation: Prepare precursor solution containing Fe³⁺ and [PtCl₆]²⁻ in glycol solvent to enhance affinity between the hetero-electric metal groups.
- Chelation: Mix pretreated bimetallic cations with CNF‑NH₂ during freeze-drying, allowing metals to chelate stably with amine groups.
- Pyrolysis: Heat at 700°C in inert atmosphere to form the final atomic-scale bimetal assembly catalyst.
Characterization Checklist:
- STEM-HAADF: Confirm atomic dispersion and measure Pt-Fe distances (expect 2.83-2.91 Å)
- XRD: Check absence of crystalline metal nanoparticles (dominant peak should be ~25° for carbon)
- XPS/ICP-OES: Verify elemental composition and oxidation states
- EDX Mapping: Confirm uniform distribution of Pt, Fe, N, and C
Synthesis Protocol: Ni-Fe Molecular Complex via In Situ Electrochemical Activation
This protocol creates dynamic bimetallic sites through electrochemical transformation for enhanced oxygen evolution reaction [1].
Materials:
- Graphene oxide (GO) aqueous suspension
- Ni vessel
- High-purity KOH electrolyte
- Fe ion source (e.g., Fe(NO₃)₃)
- Argon atmosphere for thermal annealing
Step-by-Step Procedure:
- Prepare Ni Single-Atom Pre-catalyst:
- Seal GO suspension in Ni vessel at 80°C to assemble 3D Ni(OH)₂/graphene hydrogel
- Freeze-dry and thermally anneal at 700°C under Ar to form Ni/NiO nanoparticles and holey graphene nanomesh
- Acid treatment to yield Ni single atoms trapped in graphene nanomesh (Ni-SAs@GNM)
- Electrochemical Activation:
- Load Ni-SAs@GNM onto glassy carbon working electrode
- Use standard three-electrode system in purified 1 M KOH with 1 ppm Fe ions added
- Perform cyclic voltammetry between 1.1 and 1.65 V vs. RHE until activation complete
- Alternative: Use anodic chronopotentiometry or chronoamperometry
Characterization Checklist:
- HAADF-STEM: Verify atomic dispersion of both Ni and Fe
- Operando XAFS: Monitor structural transformation from Ni monomer to O-bridged Ni-Fe₂ trimer
- SXRF Spectroscopy: Confirm incorporation of Fe species and determine Ni/Fe ratio (~5.2:1)
- XRD/HRTEM: Rule out nanoparticle formation
Performance Benchmarking Data
Table 1: Quantitative Performance Metrics of Bimetal Assembly Catalysts
| Catalyst Material | Reaction | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| Pt = N₂ = Fe ABA | ORR | Kinetic current density (0.95 V) | 5.83 mA cm⁻² | [34] |
| Pt = N₂ = Fe ABA | ORR | 4-electron pathway selectivity | ~99% | [34] |
| Pt = N₂ = Fe ABA | Zinc-air battery | Peak power density | 198.4 mW cm⁻² | [34] |
| Commercial Pt/C | ORR | Kinetic current density (0.95 V) | ~0.06 mA cm⁻² | [34] |
| Commercial Pt/C | Zinc-air battery | Peak power density | 172.1 mW cm⁻² | [34] |
Table 2: Structural Parameters and Their Catalytic Implications
| Structural Feature | Characterization Technique | Optimal Range | Impact on Catalysis |
|---|---|---|---|
| Intermetallic Distance | STEM-HAADF, XAFS | 2.8–3.0 Å | Enables dual-site mechanism; distances too long favor single-site, too short favor 2e⁻ path [34] |
| Bridging Ligand | XAFS, FTIR | N-bridging preferred | Creates electronic coupling; enables formation of key M1–O–O–M2 intermediate [34] |
| Metal Composition Ratio | XPS, ICP-OES | System-dependent | Affects electronic structure; Ni/Fe ~5.2:1 showed high OER activity [1] |
| Dynamic Coordination | Operando XAFS | Observation of changes | Indicates adaptive active sites that can break scaling relationships during reaction [1] |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagents for Atomic-Scale Bimetal Assembly Synthesis
| Reagent/Material | Function | Example Application | Critical Notes |
|---|---|---|---|
| Amino-functionalized Carbon Nanoflakes (CNF‑NH₂) | Support material with anchoring sites | Chelates metal atoms in Pt= N₂=Fe assembly | Degree of functionalization crucial for metal loading; verify by FTIR [34] |
| Chloroplatinic Acid (H₂PtCl₆) | Platinum precursor | Provides Pt for ORR-active sites | Use with Fe precursor in glycol for better affinity; concentration controls loading [34] |
| Iron(III) Chloride Hexahydrate (FeCl₃·6H₂O) | Iron precursor | Creates Fe sites in bimetallic assemblies | Molar ratio to Pt affects final structure and performance [34] |
| High-Purity KOH Electrolyte | Electrochemical medium | Electrochemical activation and testing | Must be Fe-free for controlled studies; trace Fe contaminates and alters results [1] |
| Graphene Oxide (GO) | Support material for pre-catalysts | Forms 3D hydrogel structure with metals | Quality affects defect density and metal anchoring sites [1] |
Visualization of Concepts and Workflows
Diagram 1: Dual-Site Mechanism Overcoming Scaling Relationship
Diagram 2: Bimetallic Assembly Synthesis Workflow
The strategic design of atomic-scale bimetal assemblies with tailored intermetallic distances represents a paradigm shift in overcoming fundamental scaling relationships in catalysis. By implementing the synthesis protocols, troubleshooting guides, and characterization methodologies outlined in this technical support document, researchers can advance the development of next-generation catalysts with unprecedented activity and selectivity for sustainable energy applications.
Overcoming Implementation Hurdles: Troubleshooting Synergy and Stability in Novel Catalysts
Addressing Catalyst Heterogeneity and Elusive Active Site Identification
Technical Support Center
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary reasons my catalyst is losing activity over time? Catalyst deactivation is a common challenge and can stem from chemical, mechanical, or thermal causes [35].
- Chemical Poisoning: Impurities in the reactant stream (e.g., silicon, sulfur, arsenic) can bind strongly to active sites, rendering them inactive. Coke deposition (carbon-rich solids) is another common chemical poison [36] [35].
- Fouling/Masking: The physical deposition of foreign materials from the process stream can block pores and active sites on the catalyst surface [35].
- Sintering: Exposure to high temperatures can cause catalyst nanoparticles to agglomerate, drastically reducing the total active surface area. This process is often accelerated by the presence of water vapor and is typically irreversible [36] [35].
- Vapor-Solid Reactions: The catalyst can react with vapors to form an inactive surface layer or a volatile compound that leaves the reactor, leading to a loss of active material [35].
FAQ 2: Why is identifying the exact atomic structure of an active site so difficult? Active sites are often atomically dispersed and dynamic, making them elusive to characterize [37].
- Surface Heterogeneity: A catalyst surface contains many different types of sites (terraces, steps, kinks), and only a small fraction are catalytically active. Traditional bulk techniques often average signals from all sites, obscuring the signal from the crucial active sites [36].
- Transient Nature of Intermediates: Reaction intermediates, including radical species on amino acids like tyrosine or tryptophan, can be short-lived and exist in low concentrations, making them difficult to detect and assign unambiguously [37].
- Scaling Relations: The binding energies of different reaction intermediates (e.g., *O, *OOH) on a single metal site are often linearly correlated. This scaling relationship confines catalyst optimization, as strengthening the binding of one intermediate inevitably strengthens the binding of others, preventing all intermediates from having ideal energies for the reaction [16].
FAQ 3: How can we design catalysts to overcome the limitations imposed by scaling relationships? The key is to move from single-site to dual-site or multi-site mechanisms [16].
- Dual-Site Mechanism: By designing catalysts with two adjacent but distinct metal atoms at a precise distance, it is possible to bypass the formation of a scaling-relation-limited intermediate. For example, in the oxygen reduction reaction (ORR), a well-designed N-bridged Pt = N2 = Fe assembly can promote direct O–O radical breakage via a Pt–O–O–Fe intermediate, avoiding the problematic *OOH intermediate and its associated scaling relationship [16].
- Geometry Control: The intermetallic distance is critical. If too long, the single-site mechanism dominates; if too short, it may trigger undesired reaction pathways. The geometry must be tailored to enable the dual-binding mode [16].
FAQ 4: What experimental techniques can distinguish between different atomic configurations of catalysts? Advanced spectroscopy and microscopy are essential.
- Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS): This technique is sensitive to the molecular configuration of surface species. It can distinguish between "end-on" and "side-on" configurations of heterogenized dinuclear metal catalysts by their unique vibrational fingerprints, as demonstrated for Ir-based water oxidation catalysts [38].
- Multifrequency Electron Paramagnetic Resonance (EPR) Spectroscopy: EPR is powerful for detecting and identifying paramagnetic species, such as protein-based radicals (e.g., tyrosyl or tryptophanyl radicals) in metalloenzymes. It was crucial for identifying Tyr71 and Tyr236 as radical sites in cytochrome c peroxidase [37].
- High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM): This microscopy technique can directly image heavy metal atoms on supports. Intensity analysis of bright spots can confirm the presence of heteronuclear dual-atom sites (e.g., Pt and Fe) and measure their interatomic distance [16].
Troubleshooting Guides
Issue: Sudden Drop in Catalyst Activity or Selectivity
| Observation | Possible Root Cause | Corrective Actions & Characterization Techniques |
|---|---|---|
| Rapid activity loss with no change in temperature. | Chemical Poisoning. Impurities in the feed (S, Cl, As) are adsorbing to active sites [36] [35]. | 1. Analyze feed purity. 2. Use guard beds to remove impurities [35]. 3. Characterize: X-ray Photoelectron Spectroscopy (XPS) to detect poisons on the surface [35]. |
| Gradual, steady decline in activity over time. | Coking/Fouling. Blockage of pores and active sites by carbonaceous deposits or other materials [36] [35]. | 1. Regenerate catalyst through controlled oxidation (burn-off) or chemical treatment [35]. 2. Characterize: BET surface area analysis to measure surface area loss; Temperature-Programmed Oxidation (TPO) to study coke combustion [39] [35]. |
| Activity loss after a thermal excursion (overheating). | Sintering. Agglomeration of metal nanoparticles, reducing active surface area [36] [35]. | 1. Operate at lower temperatures or use dilution to control exotherms [35]. 2. Use thermal-stable supports (e.g., SiO2, ZrO2) [40]. 3. Characterize: XRD and STEM to observe particle size growth [39]. |
| Change in product distribution (selectivity). | Loss of Promoter or Site Blockage. A substance that enhances selectivity is no longer effective [40] [36]. | 1. Analyze catalyst composition (e.g., via XRF) to check for promoter loss [35]. 2. Characterize: Pulse chemisorption to measure available metal surface area and acidity [39]. |
Issue: Inability to Identify the Catalytically Active Site
| Challenge | Underlying Problem | Solutions & Advanced Techniques |
|---|---|---|
| The active site is a transient radical. | The reactive species is short-lived and exists in low concentrations, making it invisible to many techniques [37]. | 1. Use rapid-freeze techniques to trap intermediates. 2. Apply multifrequency EPR spectroscopy to detect and identify paramagnetic radical species, as used for tyrosyl radicals in enzymes [37]. |
| The catalyst contains multiple potential active sites (e.g., many Tyr/Trp residues). | Signals from inactive sites obscure the signal from the true active site [37]. | 1. Use site-directed mutagenesis. Systematically mutate candidate amino acids (Tyr, Trp) and observe the effect on the catalytic cycle and intermediate formation [37]. 2. Correlate spectroscopic data (EPR) with activity assays of mutants to pinpoint the responsible residue [37]. |
| The active site is a dinuclear complex with a specific geometry. | Standard techniques cannot distinguish between "side-on" and "end-on" configurations of two metal atoms [38]. | 1. Use DRIFTS with probe molecules (like CO). The vibrational coupling creates a unique doublet peak for "end-on" configurations, distinguishing it from "side-on" or single-atom sites [38]. 2. Complement with HAADF-STEM to image the dual-atom sites and measure interatomic distances [16] [38]. |
The Scientist's Toolkit: Essential Reagents & Materials
The following table details key materials used in the synthesis and characterization of advanced catalysts, particularly those designed to overcome scaling relationships [16].
| Research Reagent / Material | Function in Catalyst Research |
|---|---|
| Amino-functionalized Carbon Nanoflakes (CNF–NH2) | A high-surface-area support material. The amine groups (-NH2) act as anchoring sites to chelate metal cations (e.g., Pt, Fe), enabling the creation of atomically dispersed dual-metal sites [16]. |
| Metal Precursors (H2PtCl6, FeCl3) | The source of catalytic metals. They are co-deposited onto the functionalized support to create precursors to atomic-scale bimetal assemblies [16]. |
| Probe Molecules (Carbon Monoxide, CO) | Used in techniques like DRIFTS to characterize the surface structure of catalysts. The vibrational frequency of adsorbed CO is sensitive to the local atomic configuration, allowing differentiation between single-atom and dinuclear sites in different geometries [38]. |
| Oxide Supports (WO3, Fe2O3, TiO2) | Inorganic supports for heterogenizing molecular catalysts. The surface geometry and site spacing of the support (e.g., large spacing on WO3) can dictate whether an "end-on" or "side-on" dinuclear catalyst configuration is formed selectively [38]. |
| Spin Traps / Spin Labels | Chemicals used in EPR studies to detect and identify transient radical intermediates indirectly, particularly in complex systems like enzymes where direct assignment is difficult [37]. |
Experimental Protocols for Key Characterization
Protocol 1: Differentiating Catalyst Geometries using DRIFTS
- Objective: To identify whether a heterogenized dinuclear metal catalyst is in an "end-on" or "side-on" configuration [38].
- Methodology:
- Sample Preparation: Synthesize the catalyst by tethering a dinuclear metal complex (e.g., Ir-based) onto different oxide supports (WO3, Fe2O3). Use a reference sample with single-atom sites (Ir-SAC).
- DRIFTS Measurement: Place the catalyst powder in a DRIFTS cell. Flush with an inert gas (e.g., He) and reduce the sample if necessary.
- CO Probing: At room temperature, expose the catalyst to a flow of CO mixed with inert gas. CO molecules will adsorb onto the metal sites.
- Data Collection: Collect infrared spectra in the region of the C-O stretch (typically 1800-2200 cm-1).
- Expected Results & Interpretation:
- Single-Atom Catalyst (Ir-SAC): Will show a single peak for the C-O stretch.
- "Side-on" Dinuclear Catalyst: Will show a pair of singlet peaks for symmetric and asymmetric stretches.
- "End-on" Dinuclear Catalyst: Will show a characteristic doublet of peaks, with the lower-frequency peak attributed to the dangling top metal site and the higher-frequency peak to the bottom site bound to the support. This doublet is a key signature of the geometrically distinct active site [38].
Protocol 2: Identifying Elusive Protein Radicals using Multifrequency EPR
- Objective: To unambiguously assign the location of transient tyrosyl radicals in a metalloenzyme like cytochrome c peroxidase [37].
- Methodology:
- Sample Generation: Mix the ferric enzyme with a stoichiometric (e.g., 2-fold) excess of hydrogen peroxide directly in an EPR tube to initiate the catalytic cycle and form the radical intermediate. Flash-freeze in liquid nitrogen within seconds to trap the species.
- Site-Directed Mutagenesis: Prepare a series of mutant enzymes where candidate tyrosine residues (e.g., Tyr71, Tyr236) are mutated to phenylalanine (Y71F, Y236F).
- Multifrequency EPR: Record EPR spectra of both wild-type and mutant proteins at multiple frequencies (e.g., 9 GHz and 285 GHz). High-frequency EPR provides enhanced resolution.
- Data Analysis: Compare the EPR spectra of the mutants to the wild-type spectrum. The disappearance or alteration of specific spectral features in a mutant identifies the residue contributing to that signal.
- Expected Results & Interpretation: In the CcP example, the EPR signal of the tyrosyl radical was primarily eliminated in the Y71F and Y236F mutants, confirming Tyr71 and Tyr236 as the radical sites. Combining this with activity assays can then determine which radical is involved in substrate oxidation [37].
Workflow and Relationship Diagrams
Diagram 1: Catalyst Troubleshooting Workflow
Diagram 2: Overcoming Scaling Relationships
Ensuring Structural Stability Under Operational Reaction Conditions
Frequently Asked Questions
Q1: What are the most common signs of catalyst deactivation during operation? Catalyst deactivation typically manifests through several observable signs: a consistent decline in product yield, changes in selectivity toward undesired by-products, increased pressure drop across the reactor, and the need to progressively increase temperature to maintain conversion rates. Common mechanisms include sintering (particle agglomeration at high temperatures), poisoning by impurities like sulfur or chlorine, and coking (carbon deposition blocking active sites) [41].
Q2: How can I distinguish between catalyst sintering and poisoning? Distinguishing between these mechanisms requires post-reaction characterization. Sintering is indicated by a measurable increase in catalyst particle size and reduction in active surface area, typically analyzed through TEM or chemisorption. Poisoning shows specific chemical species adsorbed on active sites, identifiable via XPS or TPD. Experimentally, if activity restoration occurs after oxidative regeneration (burning off coke), coking was likely. If activity remains low after oxidation but recovers after reductive treatment (e.g., hydrogen to remove poisons), poisoning is probable [41].
Q3: What operational strategies can prevent catalyst deactivation? Preventative strategies include maintaining operating temperature within the optimal range to prevent thermal degradation and sintering, implementing feed purification systems (e.g., desulfurization units) to remove potential poisons, and using chemical additives like dispersants or scale inhibitors to prevent fouling. For exothermic reactions, ensure efficient cooling systems (jackets, internal coils) to prevent hotspot formation that accelerates deactivation [41].
Q4: How do integrative catalytic pairs (ICPs) enhance structural stability? Integrative catalytic pairs (ICPs) feature spatially adjacent, electronically coupled dual active sites that function cooperatively yet independently. This architecture distributes reactive intermediates across different sites, preventing the accumulation of deactivating species on uniform active sites. The functional differentiation within ICPs enables more efficient handling of multiple reaction intermediates simultaneously, reducing catalyst coking and degradation under demanding operational conditions [6].
Q5: What troubleshooting approach is recommended for unstable temperature control? Begin with a systematic review: verify temperature sensor calibration and reliability, check for heat exchanger fouling which impedes heat transfer, inspect control valve operation for stiction or deadband issues, and confirm controller tuning parameters are appropriate for the reaction kinetics. For exothermic reactions, ensure cooling capacity is sufficient and consider adaptive tuning for nonlinear processes where gain changes with operating conditions [41] [42].
Troubleshooting Guides
Catalyst Performance Degradation
Problem: Progressive decline in conversion efficiency and selectivity despite maintaining standard operating conditions.
| Investigation Method | Protocol | Expected Outcome |
|---|---|---|
| Temperature-Programmed Oxidation (TPO) | Heat catalyst from 25°C to 700°C at 10°C/min in 5% O₂/He flow; monitor CO₂ evolution with mass spectrometer. | Quantifies coke deposition; distinct CO₂ peaks indicate different carbon species. |
| Chemisorption Analysis | Expose 0.1g sample to 10% H₂/Ar at 50°C; measure uptake via pulse chemisorption; calculate dispersion. | 15-20% dispersion decrease indicates significant sintering; <5% change suggests minor structural change. |
| X-ray Photoelectron Spectroscopy (XPS) | Analyze catalyst surface with Al Kα radiation; focus on regions for potential poison elements (S 2p, Cl 2p). | Detect sulfur (>1 at%) or chlorine (>0.5 at%) confirms poisoning; absence suggests other mechanisms. |
Root Causes:
- Coking: Carbonaceous deposits blocking active sites, particularly common in high-temperature reducing environments.
- Sintering: Thermal degradation causing nanoparticle agglomeration, reducing active surface area.
- Poisoning: Strong chemisorption of impurity elements (S, Cl, heavy metals) irreversibly blocking active sites.
- Fouling: Physical deposition of inert materials from feed streams onto catalyst surfaces.
Solutions:
- For coking: Implement oxidative regeneration at 450-500°C with controlled oxygen concentration (1-5% in N₂).
- For sintering: Optimize operating temperature to remain below Tammann temperature of active metal; improve catalyst support interactions.
- For poisoning: Enhance feed purification; install guard beds for impurity removal; switch to poison-resistant catalyst formulations.
Reactor Temperature Instability
Problem: Oscillatory temperature behavior or uncontrolled temperature excursions during operation.
| Diagnostic Parameter | Measurement Protocol | Acceptable Range |
|---|---|---|
| Control Valve Stiction | Apply 0.5-1% incremental output changes in manual mode; observe valve position response. | <0.5% deadband; linear response to small changes. |
| Temperature Sensor Response Time | Compare with reference thermometer in fluidized sand bath at 300°C. | <5 seconds for 90% response to step change. |
| Heat Transfer Coefficient | Calculate from inlet/outlet temperatures and flow rates at steady state. | >80% of design specification; <15% deviation from baseline. |
Root Causes:
- Control valve stiction: Excessive friction in valve movement causing overshoot and cycling.
- Sensor placement/calibration: Inaccurate temperature measurement due to improper location or calibration drift.
- Insufficient heat transfer: Fouling of heat exchange surfaces or inadequate cooling capacity.
- Inappropriate controller tuning: Poorly matched PID parameters for the process dynamics.
Solutions:
- Perform valve stroke tests and repair/replace sticking control valves.
- Recalibrate temperature sensors and verify placement in representative locations.
- Implement adaptive tuning for controllers operating across different load conditions (e.g., 100% vs. 70% capacity).
- Clean heat exchange surfaces; verify cooling medium flow rates meet design specifications.
Pressure Drop Abnormalities
Problem: Increasing pressure drop across catalytic reactor bed impacting flow rates and productivity.
Diagnostic Procedure:
- Measure differential pressure at multiple points along reactor length.
- Compare current pressure profile with baseline performance data.
- Analyze feed composition for potential foulants or particulates.
- Perform visual inspection during shutdown for channeling or bed settlement.
Root Causes:
- Bed compaction: Physical settlement of catalyst particles reducing void fraction.
- Fines generation: Catalyst attrition producing small particles that block flow channels.
- Fouling: Deposit formation in bed interstices from feed impurities or side reactions.
- Maldistribution: Uneven flow distribution creating localized high-velocity zones.
Solutions:
- Replace top bed layer with structured internals to trap fines while maintaining flow.
- Install improved flow distribution devices (radial or axial distributors).
- Implement bed screening during turnaround to remove fines and reload with fresh catalyst.
- Add upstream filtration for feed streams containing particulates.
Experimental Protocols
Protocol 1: Accelerated Catalyst Stability Testing
Purpose: Evaluate catalyst structural stability under simulated operational conditions.
Materials:
- Fixed-bed reactor system with temperature control (±1°C)
- Mass flow controllers for gases (±1% full scale)
- Online GC or MS for product analysis
- Reference catalyst (50-100 mg)
Procedure:
- Load catalyst sample into reactor tube with quartz wool plugs.
- Activate catalyst in 5% H₂/Ar at 400°C for 2 hours (ramp rate: 5°C/min).
- Establish standard reaction conditions (e.g., CO₂ hydrogenation: 220°C, 20 bar, CO₂:H₂ = 1:3).
- Monitor conversion and selectivity every 2 hours for first 12 hours, then every 12 hours.
- After 100 hours time-on-stream, perform TPO analysis to quantify coke deposition.
- Characterize spent catalyst via TEM, XRD, and XPS for structural changes.
Data Interpretation: Calculate deactivation rate constant (kd) from exponential decay fitting of conversion versus time. Compare pre- and post-reaction surface area measurements. Correlation between kd and coke deposition rate indicates coking mechanism.
Protocol 2: In-situ Characterization of Structural Changes
Purpose: Monitor catalyst structural evolution under operational reaction conditions.
Materials:
- In-situ/reactor cell compatible with characterization technique
- Synchrotron beamline (XAS) or laboratory XRD with environmental chamber
- Gas delivery system with switching capabilities
- Reference standard (e.g., Au foil for XAS energy calibration)
Procedure:
- Mount catalyst in appropriate sample holder for characterization technique.
- Establish reaction conditions matching operational parameters.
- For XAS: Collect spectra at metal absorption edge every 15-30 minutes during first 4 hours, then hourly.
- For XRD: Monitor phase changes and crystallite size evolution with time.
- Correlate structural changes with simultaneous activity measurements.
- After experiment, cool sample in reaction environment to preserve structure.
Data Analysis: Linear combination fitting of XANES spectra to quantify oxidation state changes. EXAFS Fourier transform analysis to monitor coordination number and bond distance changes. Scherrer equation application to XRD patterns for crystallite size calculation.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Integrative Catalytic Pairs (ICPs) | Dual active sites for complex reactions; overcome scaling relationships [6] | Spatially adjacent, electronically coupled sites; enables concerted multi-intermediate reactions. |
| Single-Atom Catalysts (SACs) | Isolated metal atoms; well-defined active sites, nearly 100% atom utilization [6] | Exceptional activity and selectivity; limited for reactions involving multiple intermediates. |
| Antifouling Coatings | Prevent accumulation of deposits on reactor surfaces [41] | Applied to reactor walls and heat exchangers; reduces maintenance frequency. |
| Scale Inhibitors | Chemical additives preventing precipitation and deposition [41] | Added to reactor feed streams; particularly effective for salt precipitation fouling. |
| Smart Valve Positioners | Precise control valve operation; reduce stiction and deadband [42] | Digital communication with DCS; enables fine control adjustments for stability. |
| Adaptive Tuning Algorithms | Dynamic controller optimization for nonlinear processes [42] | Automatically adjusts PID parameters based on operating conditions. |
Diagnostic Workflow and Experimental Diagrams
Catalyst Diagnostic Workflow
Stability Testing Protocol
Achieving Optimal Synergy in Plasma-Catalyst and Hybrid Systems
FAQs and Troubleshooting Guides
FAQ 1: Why does my catalyst deactivate rapidly in plasma-catalytic methane coupling, and how can I improve its stability?
Answer: Rapid catalyst deactivation in methane coupling is frequently caused by carbon deposition (coking) or sintering of active metal sites under plasma conditions. To mitigate this:
- Strategy 1: Introduce a Second Catalyst Component. Incorporating a material with hydrogenation capability, such as Pd/Al₂O₃, in a post-plasma zone can help. This catalyst hydrogenates deposited carbon species or intermediates like C₂H₂, converting them to the desired C₂H₄ and cleaning the catalyst surface. This approach has been shown to enhance C₂H₄ selectivity to 63% [43].
- Strategy 2: Optimize Reactor Configuration. Using a two-stage plasma-thermal system separates the initial activation of methane in the plasma zone from the thermal cracking of intermediates. This prevents prolonged exposure of the catalyst to the harshest plasma conditions, thereby improving longevity [43].
FAQ 2: How can I enhance the selectivity for a specific product (e.g., ethylene) in plasma-catalytic reactions?
Answer: Achieving high selectivity requires steering reaction pathways, which can be done by:
- Strategy 1: Precise Control of Residence Time. In a two-stage plasma-thermal system for methane to ethylene conversion, the residence time in the thermal cracking stage is critical. Research indicates that an optimal residence time of ~0.14 s in the thermal reactor can shift the product distribution, increasing C₂H₄ selectivity to 68% while suppressing C₂H₆ and C₂H₂ formation [43].
- Strategy 2: Strategic Catalyst Selection. The choice of catalyst directly influences the reaction pathway. For example, in a dielectric barrier discharge (DBD) plasma, the presence of a catalyst like Pt/CeO₂-SAC can direct selectivity towards C₂ species, although the primary product may be ethane. The catalyst's role is to provide alternative, lower-energy pathways for the formation of the desired product [43].
FAQ 3: What are the primary methods for characterizing and diagnosing issues in a plasma-catalyst system?
Answer: Effective diagnosis requires probing both the plasma properties and the catalyst state.
- Method 1: Catalyst Characterization. Use operando X-ray absorption fine structure (XAFS) to monitor the dynamic structural evolution of the catalyst active sites during the reaction. This technique can reveal changes in oxidation state and coordination environment, which are crucial for understanding activation and deactivation processes [1].
- Method 2: Plasma and Product Analysis. Employ product gas analysis (e.g., gas chromatography) to track conversion and selectivity. Additionally, electron paramagnetic resonance (EPR) spectroscopy with spin trapping agents (e.g., DMPO) can be used to identify and quantify reactive radical species (like •OH) generated in the plasma, providing insight into the dominant reaction mechanisms [32].
FAQ 4: How can I overcome fundamental scaling relationships that limit catalyst activity in multi-step reactions?
Answer: Scaling relationships, where the adsorption energies of different reaction intermediates are linearly correlated, pose a fundamental limit. Innovative strategies to circumvent them include:
- Strategy 1: Employ Dynamic Dual-Site Catalysts. Design catalysts where active sites can dynamically reconfigure. For instance, a Ni-Fe molecular catalyst was shown to undergo dynamic coordination evolution during the oxygen evolution reaction, where intramolecular proton transfer alters the electronic structure of an adjacent Fe site. This simultaneous optimization of multiple intermediate adsorption energies broke the scaling relationship [1].
- Strategy 2: Utilize Dual-Atom Catalysts (DACs). DACs, particularly heteronuclear pairs (e.g., Fe-Ni), provide two adjacent but distinct metal atoms. This allows for a more flexible adsorption environment, enabling unconventional reaction mechanisms like the O-O coupling mechanism (OCM) that bypass the formation of a scaling-relation-limited intermediate (*OOH), leading to a lower overpotential [44].
- Strategy 3: Implement Spatial Confinement. Confining catalysts at the angstrom scale, such as by intercalating iron oxyfluoride (FeOF) between graphene oxide layers, can physically restrict intermediates and leached ions. This confinement preserves the catalyst's active structure and enhances its stability, effectively maintaining its high reactivity over time [32].
The table below consolidates key performance metrics from experimental studies on plasma-catalytic and hybrid systems.
Table 1: Performance Metrics in Plasma-Catalytic Processes
| Process & System Description | Key Performance Metric | Value Achieved | Key Parameter Influence | Citation |
|---|---|---|---|---|
| NOCM in Two-Stage Plasma-Thermal System | CH₄ Conversion | 32% | Optimization of thermal reactor residence time | [43] |
| C₂H₄ Selectivity | 68% | Optimization of thermal reactor residence time | [43] | |
| H₂ Selectivity | 63% | Optimization of thermal reactor residence time | [43] | |
| H₂O₂ Activation with FeOF/Graphene Oxide Membrane | Pollutant Removal Duration | >2 weeks (near-complete removal) | Spatial confinement in angstrom-scale channels | [32] |
| Radical Generation (vs. FeOCl) | 4.7x higher DMPO–OH signal | Intrinsic property of FeOF catalyst | [32] |
Detailed Experimental Protocols
Protocol 1: Synthesis and Evaluation of a Spatial Confinement Catalytic Membrane
This protocol is adapted from studies on overcoming the reactivity-stability challenge in water treatment catalysts [32].
Objective: To fabricate a catalytic membrane with enhanced stability for radical-based oxidation processes and evaluate its long-term performance.
Materials:
- Catalyst Precursor: Iron fluoride trihydrate (FeF₃·3H₂O)
- Support Material: Graphene oxide (GO) suspension
- Reagents: Methanol, Hydrogen peroxide (H₂O₂), Model pollutant (e.g., Thiamethoxam)
Procedure:
- Synthesis of Iron Oxyfluoride (FeOF):
- Place FeF₃·3H₂O in an autoclave with a methanol medium.
- Heat at 220 °C for 24 hours.
- Recover the solid FeOF catalyst via filtration and drying.
- Fabrication of Catalytic Membrane:
- Intercalate the synthesized FeOF nanoparticles between layers of graphene oxide to create an assembled membrane with aligned, angstrom-scale channels (<1 nm).
- Performance Evaluation in Flow-Through System:
- Install the membrane in a flow-through filtration setup.
- Continuously feed a solution containing the model pollutant (e.g., Thiamethoxam at ppb-ppm levels) and H₂O₂.
- Monitor the pollutant removal efficiency over time using a suitable analytical method (e.g., HPLC).
- Key Metric: The system should maintain near-complete pollutant removal for extended periods (e.g., over two weeks).
- Characterization and Diagnosis:
- Use electron paramagnetic resonance (EPR) with DMPO as a spin trap to confirm •OH radical generation.
- Employ ion chromatography (IC) to measure fluoride ion leaching from the catalyst, comparing the confined membrane versus a powder suspension to demonstrate the stabilizing effect of spatial confinement.
Protocol 2: Optimizing a Two-Stage Plasma-Thermal System for Methane to Ethylene Conversion
This protocol is based on the optimization of a hybrid plasma-thermal system for non-oxidative methane coupling [43].
Objective: To maximize methane conversion and ethylene yield by independently optimizing the plasma and thermal stages.
Materials:
- Gases: High-purity Methane (CH₄)
- Plasma Reactor: Cylindrical Dielectric Barrier Discharge (DBD) reactor with variable inner diameters (e.g., 9 mm to 60 mm)
- Thermal Reactor: Tube furnace capable of high-temperature operation (up to 1350°C)
Procedure:
- Stage 1: Plasma Reactor Optimization
- Set up the DBD plasma reactor. Feed pure CH₄ at a fixed flow rate (e.g., 20 mL/min) and applied voltage (e.g., 7.5 kV).
- Systematically vary the inner diameter of the DBD reactor. For each diameter, measure the CH₄ conversion and product distribution (C₂H₆, C₂H₄, C₂H₂, H₂).
- Identify the optimal diameter that provides a balance between conversion and selectivity towards C₂H₆ (the primary C₂ product from the plasma stage).
- Stage 2: Thermal Cracking Reactor Optimization
- Connect the outlet of the optimized plasma reactor to the inlet of the thermal cracking reactor.
- With the plasma stage active, systematically increase the temperature of the thermal reactor from room temperature up to 1350°C.
- At each temperature, analyze the product gas composition to determine the selectivity shift from C₂H₆ to C₂H₄ and C₂H₂.
- Determination of Optimal Residence Time:
- The residence time in the thermal reactor is a function of its dimensions and the gas flow rate.
- Correlate the residence time in the thermal reactor with the observed C₂H₄ selectivity. The study identified an optimal residence time of ~0.14 s at 1350°C for maximal C₂H₄ yield [43].
- Pathway Analysis:
- By analyzing the product distribution at the outlets of both stages independently, the primary reaction pathway (Plasma: CH₄ → C₂H₆ → Thermal: C₂H₆ → C₂H₄) can be confirmed.
Visualized Workflows and Strategies
Diagram 1: Strategy Framework for Overcoming Scaling Relationships
This diagram illustrates the core strategies for designing systems that circumvent fundamental catalytic limitations.
Diagram 2: Two-Stage Plasma-Thermal Reactor Workflow
This diagram outlines the experimental setup and process flow for optimized methane-to-ethylene conversion.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Plasma-Catalyst Studies
| Item | Function / Application | Specific Example(s) | Key Consideration |
|---|---|---|---|
| Catalyst Supports | Provides a high-surface-area anchor for active metal sites, can influence electronic structure. | N-doped Graphene, Graphene Oxide (GO), CeO₂, Al₂O₃ | N-doping on graphene creates strong metal-support interactions ideal for stabilizing Single-Atom Catalysts (SACs) and Dual-Atom Catalysts (DACs) [22] [44]. |
| Metal Precursors | Source of active catalytic metal sites. | Fe, Co, Ni, Cu salts (nitrates, chlorides) | Selection is based on the target reaction. Ni and Fe are common for methane reforming, while Co and Fe are prominent for oxygen reactions [1] [44]. |
| Spin Trap Agents | Used in EPR spectroscopy to detect and identify short-lived radical species generated in plasma. | DMPO (5,5-dimethyl-1-pyrroline N-oxide) | Essential for providing experimental proof of radical-driven reaction mechanisms (e.g., •OH generation) [32]. |
| Dielectric Materials | Forms the barrier in Dielectric Barrier Discharge (DBD) plasma reactors, essential for generating non-thermal plasma. | Quartz, Alumina (Al₂O₃) | The material and thickness of the dielectric barrier directly affect the discharge properties and micro-discharge distribution [43]. |
| Reference Catalysts | Benchmark materials for comparing and validating the performance of newly developed catalysts. | IrO₂, RuO₂ (for OER), Pt/C (for HER), conventional FeOCl (for AOPs) | Provides a baseline to demonstrate performance enhancement (e.g., higher activity/stability) over existing materials [32] [1]. |
Balancing Geometric and Electronic Configuration in Dual-Site Designs
Theoretical Foundation: Overcoming Scaling Relationships
FAQ: What are the fundamental advantages of a dual-site design over a conventional single-site catalyst?
In multi-step catalytic reactions, such as the Oxygen Evolution Reaction (OER) or CO₂ hydrogenation, the performance of conventional single-site catalysts is intrinsically limited by Linear Scaling Relationships (LSRs). On a single active site, the adsorption energies of different reactive intermediates (e.g., *OH, *O, *OOH in OER) are linearly correlated. This makes it thermodynamically impossible to independently optimize the binding strength of all intermediates to achieve a minimal overall energy barrier for the reaction [1] [45].
Dual-site catalysts (DSCs) circumvent this limitation by employing two spatially close but functionally distinct active sites. The core principle is dynamic cooperation:
- Geometric Configuration: The precise spatial arrangement of the two sites creates a unique micro-environment. This can involve one metal site acting as the primary adsorption site while an adjacent site, with a different electronic structure, stabilizes a specific reaction intermediate through a second-sphere interaction [1].
- Electronic Configuration: The electronic interaction between the two metal sites, often through a bridging ligand or the support, allows for a dynamic electronic structure that shifts during the catalytic cycle. This dynamic regulation can simultaneously lower the free energy of mutually competitive steps, such as O–H bond cleavage and O–O bond formation in OER, thereby breaking the scaling relationship [1] [46].
Experimental Protocols for Dual-Site Catalyst Synthesis & Characterization
This section provides detailed methodologies for creating and validating dual-site catalysts, as referenced in the provided search results.
Protocol 1: In Situ Electrochemical Activation for Molecular Complex Catalysts
Based on the synthesis of a Ni-Fe molecular catalyst for OER [1].
Objective: To construct a well-defined Ni-Fe₂ molecular complex catalyst via electrochemical activation of a single-atom pre-catalyst.
Materials:
- Pre-catalyst: Ni single atoms on a conductive support (e.g., Ni-SAs@GNM - Ni single atoms on a graphene nanomesh).
- Electrolyte: Purified 1 M KOH, with a deliberate addition of 1 part-per-million (ppm) Fe ions (from Fe salts like Fe(NO₃)₃).
- Equipment: Standard three-electrode electrochemical setup (Glassy Carbon Working Electrode, Pt counter electrode, Reference Electrode).
Procedure:
- Preparation: Load the Ni single-atom pre-catalyst onto the glassy carbon working electrode.
- Activation: Perform electrochemical activation using Cyclic Voltammetry (CV). Scan the potential between 1.1 V and 1.65 V (vs. RHE) for multiple cycles in the Fe-doped KOH electrolyte.
- Alternative: Anodic chronopotentiometry or chronoamperometry can also be used.
- Mechanism: During activation, negatively charged Fe(OH)₄⁻ anions in the electrolyte are electrically driven to the anode. They preferentially anchor onto the positively charged Ni single-atom sites, forming an oxygen-bridged Ni-Fe₂ trimer structure, which is the active dual-site catalyst.
Validation Technique: Operando X-ray Absorption Fine Structure (XAFS)
- Purpose: To probe the local coordination and dynamic structural evolution of Ni/Fe atoms during the electrochemical reaction.
- Method: Collect Ni K-edge and Fe K-edge XAFS spectra while the catalyst is under operating conditions (in situ). This confirms the transformation from Ni monomer to the O-bridged Ni-Fe₂ trimer and provides data on oxidation states and coordination numbers [1].
Protocol 2: Workflow for Machine Learning-Accelerated Screening of Inverse Catalysts
Based on the exploration of InᵧOₓ/Cu(111) for CO₂ hydrogenation [46].
Objective: To efficiently discover and analyze transition states for elementary reaction steps across a wide variety of complex active sites on inverse catalysts.
Materials & Computational Setup:
- Software: Density Functional Theory (DFT) code (e.g., GPAW), Atomic Simulation Environment (ASE), Machine Learning Interatomic Potential (MLIP) package (e.g., Apax with Gaussian Moment Neural Network architecture).
- Model System: A defined inverse catalyst structure, e.g., nanoclusters of indium oxide (InᵧOₓ) supported on a Cu(111) metal slab.
Procedure:
- Generate Training Data: Use DFT to optimize a diverse set of initial structures, including the clean catalyst, adsorbates (e.g., *CO₂, *H), and potential transition state guesses. Use lower-level DFT settings (e.g., Γ-point only, 400 eV cutoff) to speed up this initial data collection.
- Train MLIP: Train a Machine Learning Interatomic Potential (e.g., a Gaussian Moment Neural Network) on the collected DFT data. The MLIP learns to predict the system's energy and atomic forces at a fraction of the computational cost of DFT.
- High-Throughput Transition State Search: Use the trained MLIP to perform rapid nudged elastic band (NEB) or dimer method calculations to locate transition states for the reaction of interest (e.g., formate formation) across dozens of different active site motifs on the inverse catalyst clusters.
- DFT Validation: Refine the most promising MLIP-predicted transition state structures using higher-level DFT calculations (e.g., with a 600 eV cutoff and a 2x2x1 k-point grid) to ensure accuracy.
- Analysis: Analyze the final dataset of activation energies and geometries to identify structure-activity trends and locate sites that break conventional scaling relations.
Table 1: Key Research Reagent Solutions for Featured Experiments
| Item | Function | Example from Research |
|---|---|---|
| Single-Atom Pre-catalyst (e.g., Ni-SAs@GNM) | Provides isolated, well-defined anchoring points for the construction of molecular complex catalysts. | Ni single atoms on graphene nanomesh [1]. |
| Fe-doped Alkaline Electrolyte | Source of secondary metal (Fe) for the in-situ electrochemical construction of the dual-site active center. | 1 M KOH with 1 ppm Fe ions [1]. |
| Inverse Catalyst Model (e.g., InᵧOₓ/Cu(111)) | A complex model system where metal oxide nanoclusters on a metal support create numerous potential dual-site motifs for screening. | Nanoclusters of indium oxide on a copper surface [46]. |
| Machine Learning Interatomic Potential (MLIP) | Enables computationally tractable exploration of reaction pathways on complex catalysts by approximating DFT-level accuracy. | Gaussian Moment Neural Network (GM-NN) potential [46]. |
Troubleshooting Guides for Dual-Site Catalyst Experiments
Problem Area 1: Poor Catalytic Activity & Selectivity
Observed Symptom: Lower-than-expected conversion rate, poor product selectivity, or a failure to surpass the performance benchmark set by scaling relationship predictions.
Diagnostic Steps & Solutions:
| Symptom | Potential Cause | Investigation & Corrective Action |
|---|---|---|
| Gradual decline in conversion and selectivity. | Catalyst sintering (agglomeration of particles) leading to loss of active dual-site structures [26]. | Check: Perform post-reaction TEM/HAADF-STEM to observe particle size. Fix: Lower operating temperature to reduce thermal degradation; ensure support material has high thermal stability. |
| Rapid activity loss and unwanted side products (e.g., high gas production). | Preferential triggering of undesired side reactions (e.g., hydrocracking over dehydrocyclization) due to improper geometric configuration or feed quality [27]. | Check: Analyze feed composition (e.g., paraffin content). Fix: Adjust feed quality (e.g., use naphtha blending to control paraffin content); optimize reactor severity (temperature/pressure). |
| Failure to break scaling relations, minimal performance gain. | Ineffective cooperation between the two sites due to suboptimal distance or electronic coupling. | Check: Use operando XAFS and DFT/AIMD simulations to study the dynamic coordination environment during reaction [1]. Fix: Re-design synthesis to control the atomic spacing between metal sites; explore different metal pairings or bridging ligands. |
Problem Area 2: Catalyst Instability & Deactivation
Observed Symptom: A steady or rapid loss of catalytic activity over time, increased pressure drop across the reactor, or physical breakdown of the catalyst material.
Diagnostic Steps & Solutions:
| Symptom | Potential Cause | Investigation & Corrective Action |
|---|---|---|
| Increased reactor pressure drop (ΔP). | Coking/Carbon laydown blocking pores and flow channels, or mechanical fouling by heavy metals [26]. | Check: Analyze radial temperature profiles (variations >6-10°C indicate channeling). Monitor for catalyst fines in effluent. Fix: For coking, adjust operating conditions to lower coking tendency; implement a regeneration cycle. For fouling, improve feed pre-treatment. |
| Low reactor ΔP and poor conversion. | Channeling due to poor initial catalyst loading, creating voids and flow bypassing [26]. | Check: Confirm with radial temperature measurements. Fix: Ensure proper catalyst loading procedures are followed. A reactor re-load might be necessary. |
| Loss of active sites, decreased surface area. | Chemical poisoning by feed impurities (e.g., S, Cl) strongly chemisorbing on active sites [26]. | Check: Implement rigorous feed impurity monitoring (S, Cl content). Fix: Improve feed purification pre-treatment; consider using a guard bed before the main reactor. |
Problem Area 3: Inconsistent Experimental Results
Observed Symptom: Poor reproducibility between catalyst batches, or erratic performance data during the same experimental run.
Diagnostic Steps & Solutions:
- Cause: Maldistribution of Reactant Flow. Uneven flow across the catalyst bed can create local hot/cold spots and varying residence times, leading to erratic temperature profiles and product yields [26].
- Solution: Inspect and clean the reactor's inlet distributor. Confirm uniform catalyst packing to prevent channeling.
- Cause: Improper Water/Chloride Balance (for reforming catalysts). The balance between the catalyst's acidic and metal functions is critical. An incorrect water-to-chloride molar ratio can lead to a loss of activity and unpredictable performance [27].
- Solution: Monitor and control the water to chloride molar ratio in the recycle gas (typically to a range of 15-25 for fixed-bed reformers, as per licensor specifications) [27].
- Cause: Uncontrolled Interaction Effects in Parallel Experiments. If running multiple related experiments (e.g., on the same flow or page), visitors or data points exposed to multiple experimental conditions can behave differently, skewing results [47].
- Solution: For physical experiments, ensure strict isolation of variables. For computational or high-throughput screening, use a Sequential Design of Experiments (SDOE) approach. SDOE uses results from early batches to inform the choice of subsequent experiments, maximizing learning and reducing noise from exploring non-optimal conditions [48].
Visualization of Workflows
The following diagrams illustrate the core experimental and computational workflows for developing dual-site catalysts.
Diagram 1: Dual-Site Catalyst Development Workflow. This chart outlines the iterative process from theoretical design to experimental validation and troubleshooting.
Diagram 2: ML-Accelerated Screening for Inverse Catalysts. This workflow demonstrates how machine learning interatomic potentials can be used to efficiently discover new active sites that break linear scaling relationships [46].
Table 2: Key Catalyst Characterization Techniques
| Technique | Acronym | Key Information Provided | Relevance to Dual-Site Catalysts |
|---|---|---|---|
| X-ray Absorption Fine Structure | XAFS | Local electronic structure, oxidation state, coordination number, interatomic distances. | Crucial for confirming the presence of two distinct metal sites and their coordination environment. Operando XAFS tracks dynamic changes [1]. |
| Aberration-Corrected High-Angle Annular Dark-Field Scanning TEM | AC-HAADF-STEM | Atomic-resolution imaging of heavy atoms on lighter supports. | Directly visualizes the atomic dispersion of metal atoms and can confirm the co-existence of two different metals in close proximity [1]. |
| Density Functional Theory + Ab Initio Molecular Dynamics | DFT + AIMD | Energetics of reaction pathways and simulation of dynamic site evolution at finite temperature. | Models the dynamic cooperation between sites and identifies transition states, providing the theoretical basis for broken scaling relations [1]. |
Mitigating Metal Leaching and Deactivation in Bimetallic Systems
A technical guide for catalyst stabilization in advanced intermediates research
Troubleshooting FAQs: Metal Leaching & Catalyst Deactivation
Q1: What are the primary causes of sudden activity loss in my bimetallic catalyst during reaction cycles?
The most common causes fall into three categories, each with distinct diagnostic characteristics:
- Metal Leaching: The active metal components dissolve into the reaction medium. This is confirmed via Inductively Coupled Plasma Mass Spectrometry (ICP-MS) analysis of the reaction solution post-reaction, showing a measurable decrease in metal content on the catalyst support [49].
- Carbon Deposition (Coking): Polycyclic aromatic hydrocarbons or other carbonaceous species block active sites and pores. This is identified through Thermogravimetric Analysis (TGA), which shows mass loss at high temperatures in an oxygen-rich atmosphere as the carbon burns off [50] [51].
- Metal Sintering/Particle Growth: Nanoparticles agglomerate into larger particles, reducing the total active surface area. This is detected via Transmission Electron Microscopy (TEM) and X-ray Diffraction (XRD), which show an increase in the average particle size and sharper diffraction peaks, respectively [49] [51].
Q2: My catalyst deactivates rapidly due to coking. What strategies can I implement to reduce carbon formation?
Adjusting the properties of the active metal and its support can significantly suppress coke formation.
- Optimize Metal-Support Interaction: Choose a support that strongly anchors metal nanoparticles. A strong interaction prevents metal migration and particle agglomeration at high temperatures, which are precursors to filamentous carbon formation [51].
- Utilize Bimetallic Synergy: Incorporate a second metal that promotes the gasification of carbon precursors. For example, in Ni-based catalysts for dry reforming of methane (DRM), adding Sn, Au, or Pt can break larger Ni ensembles that are active for carbon formation [51].
- Tune Surface Acidity/Basicity: Basic supports or additives can chemically adsorb and activate CO₂, which then helps to oxidize and remove surface carbon deposits as CO [51].
Q3: I suspect metal leaching is occurring in my liquid-phase reaction. How can I confirm this and what are my options?
Confirmation and mitigation strategies for metal leaching are as follows:
- Confirmation: Perform ICP-MS on the cooled reaction solution after separating the catalyst. Compare the metal concentrations with a fresh reaction mixture to quantify leaching [49].
- Mitigation Strategies:
- Strengthen Metal-Support Interaction: Employ synthesis methods that create strong electronic or covalent bonds between the metal nanoparticles and the functional groups on the support surface [49] [51].
- Modify the Reaction Environment: Avoid highly acidic reaction conditions that promote dissolution. If possible, use neutral or basic media. The addition of corrosion inhibitors can also be effective in some aqueous systems [52].
- Alloying: Forming a stable alloy can protect the more leachable component. For instance, in Pd-Ag catalysts, the presence of Pd can help regenerate the chlorinated silver surface, slowing deactivation [53].
Q4: Can a deactivated catalyst be regenerated, and what is a standard protocol?
Yes, many forms of deactivation are reversible. The regeneration protocol depends on the deactivation mechanism. A common method for coke removal is controlled combustion [50] [54].
- Protocol for Regeneration via Coke Combustion:
- Purge: After reaction, purge the reactor with an inert gas (e.g., N₂) to remove any residual flammable gases.
- Oxidative Calcination: Introduce a dilute stream of O₂ (e.g., 2-5% in N₂) and slowly ramp the temperature (e.g., 2-5°C/min) to a target between 450-550°C. Hold at this temperature for 2-4 hours. The low O₂ concentration prevents excessive exothermicity that could damage the catalyst [50].
- Reduction (if needed): For metal oxides formed during calcination, follow with a reduction step in a H₂ stream (e.g., 5% H₂ in N₂) at a suitable temperature (e.g., 400-500°C) for 1-2 hours to reduce the active metal back to its metallic state [49].
- Cool and Purge: Cool the catalyst to reaction temperature under an inert atmosphere.
Table 1: Common Catalyst Deactivation Mechanisms and Diagnostic Methods
| Deactivation Mechanism | Primary Diagnostic Techniques | Key Observations |
|---|---|---|
| Metal Leaching [49] | ICP-MS | Decreased metal loading on spent catalyst; metal ions in solution. |
| Carbon Deposition (Coking) [50] [51] | TGA, TEM | Mass loss in TGA; visible carbon filaments or layers in TEM. |
| Metal Sintering [49] [51] | TEM, XRD | Increased metal particle size; sharper XRD peaks due to larger crystals. |
| Active Site Poisoning [51] | XPS, Chemisorption | Presence of foreign elements (e.g., S) on surface; loss of metal surface area. |
Table 2: Regeneration Techniques for Different Deactivation Types
| Deactivation Type | Regeneration Method | Process Conditions | Limitations |
|---|---|---|---|
| Coke Deposition [50] [54] | Controlled Combustion | 450-550°C, 2-5% O₂ in N₂ | Can cause sintering if temperature is too high. |
| Reversible Oxidation [49] | Reduction | 400-500°C, 5% H₂ in N₂ | May not restore original dispersion if sintering occurred. |
| Surface Contamination [49] | Solvent Washing | Washing with methanol or other solvents. | Less effective for strongly chemisorbed poisons or internal coke. |
Experimental Protocols for Deactivation Analysis
Protocol 1: Assessing Metal Leaching in Liquid-Phase Reactions
This protocol is essential for quantifying the loss of active metal species during reaction.
- Catalyst Separation: After the reaction is complete and the reactor has cooled, separate the solid catalyst from the reaction mixture by centrifugation or filtration (using a filter with pores <0.45 µm).
- Digestion of Spent Catalyst: Accurately weigh a portion of the recovered, dried spent catalyst. Digest it in a mixture of concentrated nitric and hydrochloric acid (aqua regia) using a microwave digester to ensure complete dissolution of metal particles.
- Solution Analysis: Dilute the digested solution and the separated reaction filtrate to appropriate volumes with deionized water. Analyze both solutions using ICP-MS to determine the metal concentrations.
- Calculation: The extent of leaching is calculated by comparing the metal content in the spent catalyst with a fresh catalyst, and by confirming the presence of leached metal in the reaction filtrate [49].
Protocol 2: Quantifying Coke Deposition via Thermogravimetric Analysis (TGA)
This method measures the amount of carbonaceous deposit on a spent catalyst.
- Sample Preparation: Place 10-20 mg of the spent catalyst in a TGA crucible. Ensure the sample is dry.
- Temperature Program:
- Step 1 (Purge): Ramp from room temperature to 150°C at 10°C/min under a N₂ atmosphere (50 mL/min flow) and hold for 10 minutes to remove moisture.
- Step 2 (Combustion): Switch the gas from N₂ to synthetic air (20% O₂ in N₂). Ramp the temperature to 800°C at 10°C/min and hold for 20-30 minutes. The mass loss in this step corresponds to the combustion of coke.
- Data Analysis: The percentage mass loss during the combustion step (Step 2) is reported as the weight percent of coke on the catalyst [50].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Catalyst Deactivation Studies
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| ZSM-5 Zeolite Support [50] | Microporous solid acid support; provides shape selectivity and acid sites for reactions like catalytic pyrolysis. | Used as a support for Fe-Ni bimetallic catalysts to produce aromatics [50]. |
| MgO-Modified Al₂O₃ Support [49] | Basic support; helps neutralize acid sites that promote coking and can strengthen metal-support interaction. | Used for Pd-Pb catalysts in oxidative esterification to improve stability [49]. |
| Hydrazine Solution (Aqueous) [49] | Mild reducing agent; used to reduce surface oxides on metal nanoparticles during regeneration. | Effective for regenerating deactivated Pd-Pb catalysts without causing sintering [49]. |
| Lactobacillus plantarum [55] | Bioleaching agent; produces organic acids that can selectively leach metals from waste streams or contaminated catalysts. | Used for sustainable recovery of metals from sludge via bioleaching [55]. |
| h-BN (Hexagonal Boron Nitride) Support [22] | 2D support material with high thermal stability and tunable defects; can anchor single-atom or dual-atom catalysts. | Serves as a substrate for Fe-Ni dual-atom catalysts to break scaling relationships in CO₂ reduction [22]. |
Experimental Workflow & Catalyst Regeneration Pathway
The following diagram illustrates a logical pathway for diagnosing and addressing catalyst deactivation, integrating the FAQs and protocols above.
Diagram 1: Catalyst Deactivation Troubleshooting Workflow
Advanced Context: Linking to Scaling Relationships
The challenge of mitigating metal leaching and sintering is intrinsically linked to the broader thesis of overcoming linear scaling relationships in catalysis. Traditional single-atom or uniform active sites often face a fundamental limitation: the adsorption energies of different reaction intermediates are linearly correlated, placing a ceiling on catalytic performance [6] [22].
The use of bimetallic systems and integrative catalytic pairs (ICPs) presents a strategic path forward. As explored in the troubleshooting guide, a well-designed bimetallic catalyst like Fe-Ni or Pd-Pb does more than just resist deactivation. The synergistic interaction between two different, adjacent metal atoms creates dual active sites that can function cooperatively yet independently [6]. This allows for the independent tuning of adsorption strengths for multiple intermediates simultaneously, thereby breaking the constraining linear scaling relationships [22]. Consequently, a catalyst designed for stability against leaching and sintering, through strong metal-support interaction and optimal alloying, can also achieve superior activity and selectivity by its very nature as an integrative catalytic pair.
Validating Success: Comparative Analysis of Catalytic Performance and Mechanistic Evidence
A fundamental challenge in the rational design of advanced catalysts is overcoming the limitations imposed by linear scaling relationships (LSRs). These relationships create inherent constraints on simultaneously optimizing the binding energies of multiple reaction intermediates on conventional single-site catalysts. For multi-step reactions like the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), LSRs inevitably place an upper limit on catalytic performance [16] [11]. Operando X-ray Absorption Fine Structure (XAFS) spectroscopy, which includes both Extended XAFS (EXAFS) and X-ray Absorption Near Edge Structure (XANES), has emerged as a powerful technique to probe the dynamic local electronic and geometric structures of catalytic active sites under working conditions. By revealing how catalyst structures dynamically evolve in response to reaction environments, operando XAFS provides the critical insights needed to design strategies that circumvent these scaling relationships, such as dual-site mechanisms that bypass unfavorable intermediates [16] [11].
This technical support center provides essential troubleshooting guides and FAQs to help researchers obtain high-quality, interpretable data from their operando XAFS experiments, directly enabling the study of dynamic catalytic processes.
Core Concepts: XAFS and its Application to Catalysis
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between XANES and EXAFS, and what specific information does each provide? XAFS is divided into two primary regions that deliver complementary information:
- XANES (X-ray Absorption Near Edge Structure): This region encompasses the spectrum from about 50 eV below the absorption edge to approximately 50-100 eV above it. It is highly sensitive to the local electronic structure, including the oxidation state and coordination geometry (e.g., octahedral, tetrahedral) of the absorbing atom. The edge position shifts to higher energy with increasing oxidation state [56].
- EXAFS (Extended X-ray Absorption Fine Structure): This is the oscillatory part of the spectrum extending from about 50-100 eV above the edge to higher energies. EXAFS analysis provides quantitative information about the local coordination environment, including interatomic distances, coordination numbers, and disorder (Debye-Waller factor) of the shells of atoms surrounding the absorber [56].
Q2: What does 'operando' specifically mean in the context of XAFS experiments, and why is it crucial for catalysis research?
- In-situ measurements are performed on a catalytic system under simulated reaction conditions (e.g., elevated temperature, applied voltage, presence of reactants).
- Operando measurements are a subset of in-situ studies where the catalyst is probed under working conditions while simultaneously measuring its catalytic activity (e.g., conversion, selectivity). This direct correlation between structure and function is essential for establishing genuine structure-activity relationships and identifying the true nature of the active site, which can differ significantly from its static, pre-catalytic form [57] [11].
Q3: How can operando XAFS specifically help in overcoming linear scaling relationships in catalysis? Operando XAFS can directly observe and verify the formation of novel active sites that operate via mechanisms different from the conventional single-site pathway. For instance:
- It can confirm the existence of a dual-site mechanism, where two adjacent metal atoms break the O-O bond in ORR without forming the *OOH intermediate, thereby bypassing the scaling relationship that governs the single-site mechanism [16].
- It can track the dynamic evolution of active sites, such as the transformation of a Ni single-atom pre-catalyst into an O-bridged Ni-Fe2 trimer during OER. This dynamic coordination can modulate the electronic structure of the active center during the catalytic cycle, simultaneously optimizing the energetics of multiple steps and breaking the scaling relationship [11].
Experimental Protocols & Methodologies
A Standard Workflow for Operando XAFS in Electrocatalysis
The following diagram outlines a generalized workflow for conducting an operando XAFS study on an electrocatalytic system, such as the oxygen evolution reaction (OER).
Diagram 1: Operando XAFS experimental workflow for electrocatalysis.
Step-by-Step Protocol:
- Catalyst Synthesis & Electrode Preparation: Synthesize the catalyst material (e.g., via impregnation [58] or molecular complex construction [11]). Prepare a homogeneous electrode by depositing a well-dispersed catalyst ink onto a conductive substrate (e.g., carbon paper or glassy carbon). The catalyst layer should be uniform to avoid artifacts.
- Operando Electrochemical Cell Assembly: Integrate the working electrode into a specialized operando cell. This cell must:
- Allow for the controlled flow of electrolyte and reactants.
- Provide electrical connections for a standard three-electrode setup (working, counter, reference).
- Feature X-ray transparent windows (e.g., Kapton, quartz capillary [58]) to allow the beam to pass through the sample.
- Enable simultaneous gas and liquid product analysis if possible [57].
- Beamline Setup & Energy Calibration: At the synchrotron beamline, configure the monochromator for the desired energy range. Calibrate the energy scale using a metal foil (e.g., Pt or Ni foil) of known absorption edge energy, measured simultaneously in transmission with the sample for accuracy.
- Simultaneous Data Acquisition: This is the core of the operando experiment.
- Begin collecting XAFS spectra at the relevant absorption edge (e.g., Pt L₃-edge for Pt catalysts).
- Simultaneously, control the electrochemical workstation to apply a potential/current and record the activity data (current, potential) and product formation.
- For time-resolved studies, use techniques like QEXAFS (Quick-scanning EXAFS) to rapidly acquire full spectra, enabling the tracking of dynamic changes on the second-to-minute timescale [58].
- Data Processing: Process the raw XAS data using standard software (e.g., Athena, Demeter). This involves energy alignment, background subtraction, normalization, and Fourier transformation of the EXAFS oscillations.
- Structural & Electronic Analysis:
- Analyze XANES spectra to track changes in oxidation state and electronic structure.
- Fit the EXAFS spectra to structural models to extract quantitative parameters (coordination numbers, distances).
- Correlate Structure with Activity: Overlay the extracted structural parameters (e.g., oxidation state, bond distance) with the simultaneously recorded electrochemical activity data to establish direct structure-activity relationships.
Research Reagent Solutions: Essential Materials for Operando XAFS
Table 1: Key materials and reagents for operando XAFS experiments in catalysis.
| Item | Function & Importance | Example from Literature |
|---|---|---|
| Catalyst Precursors | To synthesize the active catalyst material. | H₂PtCl₆·xH₂O for Pt-based catalysts [58]; Ni salts for Ni-Fe complexes [11]. |
| Carbon Support | Provides a conductive, high-surface-area substrate to anchor and disperse atomic or nanoparticle catalysts. | Graphene Oxide (GO) [11]; γ-alumina (Al₂O₃) [58]; functionalized carbon nanoflakes [16]. |
| Operando Cell Components | Enables spectroscopy under working conditions. X-ray windows are critical. | Quartz capillary microreactors [58]; cells with Kapton windows. |
| Reference Foils | Essential for accurate energy calibration of the X-ray monochromator. | High-purity metal foils (e.g., Pt, Ni, Fe). |
| Electrolyte | The medium in which the electrochemical reaction occurs. Must be pure to avoid contamination. | 1 M KOH for OER [11]; 0.1 M HClO₄ for ORR. Ultrapure grades are recommended. |
Troubleshooting Common Experimental Challenges
FAQs on Data Quality and Interpretation
Q4: My XAFS spectrum has unusual spikes or non-statistical noise. What could be the cause? Spikes or reproducible non-statistical noise are often caused by monochromator glitches or inadequate harmonic rejection.
- Solution: Suspect non-efficient harmonic rejection if spikes are present. Operate the double-crystal monochromator (DCM) with a slight detuning (e.g., 10-50%) to reduce higher-order harmonics in the X-ray beam. Ensure that harmonic-rejecting mirrors are correctly aligned. Checking the spectrum of a known standard material (like a metal foil) under the same conditions can help identify this issue [59].
Q5: The signal-to-noise ratio in my fluorescence data is poor. How can I improve it? Poor signal-to-noise (S/N) ratio in fluorescence mode is common, especially for dilute samples. The S/N ratio is given by S/√(S + B), where S is signal counts and B is background counts.
- Solution: To optimize S/N:
- Use energy-discriminating detectors (e.g., solid-state detectors) to reject scattered X-rays that contribute to the background.
- Employ X-ray filters (e.g., Z-1 filters) or Soller slits to reduce background signal.
- Ensure your sample is homogeneous and of optimal thickness. A sample that is too thick can cause self-absorption effects, distorting the spectrum [59].
Q6: How can I be sure that the structure I measure is the true active site and not a resting state? This is a central challenge in catalysis research.
- Solution:
- Perform a detailed operando experiment where you correlate the evolution of the geometric/electronic structure (from XAFS) with the actual catalytic activity (e.g., current, product formation rate).
- Look for structural parameters that change coincidentally with the onset of catalysis.
- Use time-resolved (e.g., QEXAFS) methods to capture transient intermediates [58] [11]. For example, in the study of the Ni-Fe catalyst, operando XAFS was crucial to confirm the transformation from a Ni monomer to an O-bridged Ni-Fe2 trimer during the electrochemical activation, identifying the true active structure [11].
Advanced Techniques: From Single-Point to Spatially Resolved Spectroscopy
For complex catalytic systems, the reaction environment can create spatial gradients within the reactor. A powerful advancement is the combination of rapid XAFS with full-field imaging, as illustrated in the study of a Pt/Al₂O₃ catalyst during methane CPO [58].
Diagram 2: Workflow for rapid, spatially resolved XANES imaging.
This methodology enabled the visualization of a reduction front moving through the catalyst bed, which would be impossible to detect with a conventional, single-point measurement [58]. The key quantitative aspects of such advanced setups are summarized below.
Table 2: Quantitative parameters for advanced operando XAFS setups from literature examples.
| Application / Catalyst | Technique | Time Resolution | Spatial Resolution | Key Finding |
|---|---|---|---|---|
| Pt/Al₂O₃ during CH₄ CPO [58] | Full-field QEXAFS Imaging | ~1.6 s per stack (80 images) | Micrometer-scale (full field of view) | Observation of a Pt reduction front propagating at mm/s scale during reaction ignition. |
| Ni-Fe Molecular Catalyst for OER [11] | Operando QEXAFS | Seconds to minutes per spectrum | Single-point (bulk average) | Dynamic coordination change from Ni monomer to Ni-Fe-O trimer active site. |
| Pt = N₂ = Fe for ORR [16] | In situ XAFS | Not specified (steady-state focus) | Single-point (bulk average) | Confirmation of atomic-scale Pt-Fe pair structure and direct O-O breakage mechanism. |
Electrokinetic studies provide powerful experimental tools for probing the intricate details of reaction mechanisms, particularly in the field of catalysis where understanding and overcoming linear scaling relationships (LSRs) is paramount. LSRs describe the theoretical constraint where the adsorption energies of different catalytic intermediates on a catalyst surface are linearly correlated, placing intrinsic limitations on optimally adjusting the adsorption of every intermediate simultaneously to achieve maximum activity [1] [5]. This fundamental limitation creates a "volcano plot" relationship, where catalytic performance peaks at a certain binding energy, making it impossible to continuously improve catalysts by tuning a single property. Your research aims to circumvent these scaling relationships, and electrokinetics serves as a critical methodology for identifying how dynamic structural changes in catalysts can alter the energy landscapes of multi-step reactions, thereby providing a path beyond these fundamental limitations [1].
Key Concepts and Terminology
Electrokinetic Studies combine electrochemical measurements with kinetic analysis to elucidate reaction mechanisms by determining how reaction rates depend on potential and concentration. Within catalysis research, these studies are indispensable for identifying the rate-determining step (RDS)—the slowest elementary step in a multi-step reaction mechanism that dictates the overall reaction rate [60]. The reaction mechanism itself is the detailed, step-by-step sequence of elementary reactions by which an overall chemical change occurs [61] [60]. When a catalyst's active site undergoes dynamic structural regulation, its coordination environment and electronic structure change during the catalytic cycle, which can effectively alter the adsorption energies of intermediates and potentially break conventional scaling relationships [1].
Troubleshooting Guide: Common Experimental Issues and Solutions
Interpreting Tafel Slopes and Reaction Orders
| Problem | Possible Causes | Diagnostic Tests | Solutions |
|---|---|---|---|
| Inconsistent Tafel slopes | - Change in RDS with potential- Shifting reaction mechanism- Unaccounted mass transport effects | - Measure Tafel slope at different potential ranges- Determine reaction orders at various potentials | - Verify low IR-drop and clean electrode surface- Use multiple complementary techniques to confirm mechanism [60] |
| Non-integer reaction orders | - Multi-step mechanism with pre-equilibrium- Mixed adsorption isotherms- Site-blocking co-adsorbates | - Vary reactant concentration systematically- Use isotopic labeling | - Analyze data within pre-equilibrium model framework: Rate = k Keq[A][B] if A+B⇌C (fast) followed by C→D (slow) [60] |
| Hysteresis in CV scans | - Slow structural rearrangements of catalyst- Formation/breaking of metal-adsorbate coordination- Catalyst oxidation state changes | - Operando XAFS or Raman spectroscopy- Variation of scan rate | - Incorporate dynamic structural analysis; e.g., monitor Ni-Fe coordination evolution with operando XAFS [1] |
Addressing Catalyst and System Instability
| Problem | Possible Causes | Diagnostic Tests | Solutions |
|---|---|---|---|
| Decaying current density | - Catalyst degradation/dissolution- Active site poisoning by intermediates- Uncontrolled pH changes | - ICP-MS of electrolyte- Post-mortem XPS/TEM analysis | - Introduce stabilizing ligands or supports- Implement pH-static control via buffer electrolytes |
| Unreproducible kinetics | - Trace metal contamination (e.g., Fe in KOH)- Uncontrolled electrolyte impurities- Inconsistent catalyst activation | - Ultra-purify electrolytes- Standardize pre-conditioning protocols | - Use Fe-free KOH for Ni-based OER studies; deliberately add known Fe ppm levels for reproducible Ni-Fe complex formation [1] |
| Inability to differentiate mechanisms | - Overlapping elementary step energies- Insensitive electrochemical probes | - Isotope kinetic effect measurements- Compute theoretical scaling relationships | - Combine electrokinetics with theoretical calculations (DFT/AIMD) to identify O–H cleavage vs. O–O formation barriers [1] |
Frequently Asked Questions (FAQs)
Q1: How can electrokinetic studies specifically help in breaking linear scaling relationships in catalysis? Electrokinetic studies can identify when a catalyst operates via a non-conventional mechanism that bypasses the limitations imposed by LSRs. For example, in the oxygen evolution reaction (OER), conventional catalysts exhibit a linear scaling relationship between the adsorption energies of *OOH and *OH intermediates, creating an intrinsic thermodynamic overpotential limit. Through detailed Tafel and reaction order analysis, researchers can detect dynamic dual-site mechanisms where the coordination environment of the active site evolves during the reaction. In a Ni-Fe molecular complex catalyst, electrokinetic data revealed that dynamic Ni-adsorbate coordination, driven by intramolecular proton transfer, simultaneously lowered the free energy for both O–H bond cleavage and O–O bond formation—something impossible within conventional scaling relationships [1].
Q2: What is the fundamental difference between a reaction mechanism and a rate-determining step? The reaction mechanism is the complete, step-by-step molecular-level pathway describing how reactants transform into products, including all intermediates and transition states. In contrast, the rate-determining step is the slowest elementary reaction within that sequence, which acts as a "bottleneck" controlling the overall reaction rate. The molecularity of the RDS directly determines the form of the experimental rate law. For instance, a mechanism may comprise multiple steps (e.g., a fast pre-equilibrium followed by a slow catalytic step), but the rate law will reflect only the molecularity of the slow, rate-determining step [60].
Q3: How can we experimentally distinguish between a single-site and a dual-site mechanism using electrokinetics? Dual-site mechanisms often exhibit distinctive electrochemical signatures compared to single-site pathways:
- Reaction orders: A reaction order of 2 with respect to the catalyst surface sites suggests a dual-site mechanism, whereas a first-order dependence typically indicates a single-site process.
- Tafel slopes: Unusual Tafel slopes that don't correspond to known single-electron transfer steps can indicate cooperative effects between adjacent sites.
- Kinetic isotope effects: Different KIE values can emerge when proton transfer occurs between sites versus at a single site. For the Ni-Fe OER catalyst, the combination of anomalous Tafel slopes and theoretical calculations revealed a mechanism where the Ni site dynamically coordinated to adsorbates, thereby electronically modulating the adjacent Fe active center—a clear dual-site cooperation [1].
Q4: Why is the identification of the rate-determining step so crucial for catalyst design? Identifying the RDS allows for targeted catalyst optimization because the activation energy of the RDS represents the primary kinetic barrier limiting the overall reaction rate. Once the RDS is known, catalyst design can focus on specifically stabilizing the transition state of that particular step. For example, if O–O bond formation is identified as the RDS in OER, catalysts can be designed with dual sites that optimally position oxygen species for coupling. If O–H bond cleavage is rate-limiting, catalysts with proton-acceptor functionalities can be developed. This targeted approach is far more efficient than random catalyst screening [60].
Q5: What experimental evidence confirms that a catalyst has truly broken linear scaling relationships? Definitive evidence requires a combination of electrochemical and spectroscopic data:
- Electrokinetic data showing simultaneous improvement in the kinetics of steps that are normally counter-correlated in conventional scaling relationships.
- Theoretical calculations demonstrating a deviation from the predicted scaling line between intermediate adsorption energies.
- Operando structural characterization confirming dynamic structural changes that alter the active site during catalysis. In the case of the Ni-Fe molecular catalyst, DFT calculations combined with electrokinetic studies showed that the dynamic structural regulation simultaneously lowered the free energy for both *OOH formation and *O–H bond cleavage, steps that are intrinsically linked by scaling relationships in static catalysts [1].
Essential Research Reagent Solutions
Table: Key Research Reagents for Electrokinetic Studies of Catalytic Mechanisms
| Reagent/ Material | Function in Electrokinetic Studies | Example Application & Rationale |
|---|---|---|
| Ultra-pure KOH electrolyte | Provides clean alkaline environment for OER/ORR studies without trace metal contamination | Essential for studying Ni-Fe catalysts; deliberate Fe addition (1 ppm) enables controlled in-situ formation of active Ni-Fe trimer complexes [1] |
| Isotopically labeled water (H₂¹⁸O, D₂O) | Tracing oxygen/proton pathways; Kinetic Isotope Effect (KIE) measurements | Differentiates between O–O bond formation mechanisms in OER; KIE reveals if proton transfer is rate-determining [60] |
| Fe(OH)₄⁻ species | Precursor for controlled Fe incorporation into catalyst matrix | Electrically driven to anode to form well-defined Ni-Fe molecular complexes during electrochemical activation [1] |
| Proton acceptors | Modifying proton transfer kinetics; identifying RDS | If RDS involves proton transfer, addition of proton acceptors significantly enhances reaction rate [1] |
| Permeable membranes (CEM/AEM) | Controlling ion transport; studying specific migratory species | In electrokinetic setups, CEM blocks OH⁻ from cathode, preventing precipitation of cationic intermediates for clearer kinetics [62] |
Quantitative Data in Electrokinetic Analysis
Table: Key Electrokinetic Parameters for Mechanism Differentiation
| Parameter | Significance | Typical Values for Common Mechanisms | Interpretation Guide |
|---|---|---|---|
| Tafel Slope | Indicates possible RDS based on relationship between overpotential and current | - ~120 mV/dec: 1e– RDS before chemical step- ~60 mV/dec: 1e– transfer after chemical step- ~40 mV/dec: Multi-electron transfer or dual-site | Anomalous values may signal breaking of scaling relationships via dynamic site regulation [1] |
| Reaction Order (reactant) | Molecularity of reactants in RDS | - 0: Saturated surface coverage- 1: First-order dependence- 2: Dual-site involvement | Non-integer orders suggest complex pre-equilibria; dual-site mechanisms may show order >1 [60] |
| Reaction Order (catalyst) | Number of active sites involved in RDS | - 1: Single-site mechanism- 2: Dual-site cooperative mechanism | Reaction order of 2 with respect to catalyst sites provides strong evidence for dual-site mechanism [1] |
| Transfer Coefficient (α) | Symmetry of energy barrier | 0 < α < 1; typically ~0.5 for single-electron transfer | Values deviating significantly from 0.5 may indicate changing adsorption energies during charge transfer [60] |
| Apparent Activation Energy | Temperature dependence of RDS | Typically 30–70 kJ/mol for thermally activated processes | Changes with potential if RDS shifts; lower values after catalyst modification indicate improved kinetics [1] |
Experimental Protocols for Key Electrokinetic Analyses
Protocol for Tafel Analysis and Reaction Order Determination
Objective: Determine the Tafel slope and reaction orders to identify the rate-determining step and propose a reaction mechanism.
Materials:
- Potentiostat/Galvanostat with temperature control
- Ultra-pure electrolyte (e.g., 1 M KOH, Fe-free when necessary)
- Rotating disk electrode (RDE) system to control mass transport
- Catalyst-coated working electrode, counter electrode, reference electrode
Procedure:
- Catalyst Activation: Perform electrochemical activation via cyclic voltammetry (e.g., 1.1-1.65 V vs. RHE for Ni-Fe catalysts in Fe-containing KOH) until stable CV profiles are obtained [1].
- IR Compensation: Precisely measure and compensate for solution resistance using current-interruption or positive feedback techniques.
- Steady-State Polarization: Record steady-state current at various overpotentials, ensuring equilibrium at each potential.
- Tafel Plot Construction: Plot log(current density) versus overpotential, ensuring measurements are in kinetically controlled region (typically η > 50-100 mV).
- Reaction Order Determination: Vary reactant concentration systematically while measuring current at fixed overpotential.
- Data Analysis:
- Tafel slope = dη/d(log i)
- Reaction order = d(log i)/d(log C) at constant η
Troubleshooting Tip: If Tafel plots show curvature, the RDS may be potential-dependent; analyze different potential regions separately and correlate with operando structural data [1].
Protocol for Differentiating Single-Site vs. Dual-Site Mechanisms
Objective: Experimentally distinguish between single-site and cooperative dual-site reaction mechanisms.
Materials:
- Series of catalyst samples with varying site densities (controlled during synthesis)
- Isotopically labeled reactants (e.g., D₂O for KIE studies)
- Operando spectroscopy capability (e.g., XAFS cell)
Procedure:
- Site Density Variation: Synthesize a series of catalysts with systematically varying active site densities while maintaining identical electronic structures.
- Rate vs. Site Density: Measure reaction rates normalized to geometric surface area across the site density series.
- Kinetic Isotope Effects: Measure rates with H₂O versus D₂O as proton sources.
- Operando Structural Analysis: Perform simultaneous XAFS and electrochemical measurements to detect dynamic structural changes during catalysis.
- Data Interpretation:
- Linear rate vs. site density: Single-site mechanism
- Quadratic dependence: Dual-site cooperative mechanism
- Normal KIE (kH/kD = 2-7): Proton transfer in RDS
- Inverse KIE (kH/kD < 1): H-bonding in pre-equilibrium or different mechanism
Application Example: This approach confirmed the dynamic dual-site mechanism in Ni-Fe OER catalysts, where Ni sites dynamically coordinate to adsorbates and modulate adjacent Fe sites [1].
Visualization of Concepts and Workflows
Electrokinetic Mechanism Differentiation Workflow
Breaking Scaling Relationships via Dynamic Mechanisms
In multi-step electrocatalytic reactions, such as the oxygen reduction reaction (ORR) or CO2 reduction reaction (CO2RR), linear scaling relationships (LSRs) present a fundamental bottleneck. These relationships dictate that the adsorption energies of different reactive intermediates (e.g., *OH, *O, *OOH) are correlated, making it impossible to independently optimize the binding strength of each intermediate to achieve maximal catalytic activity [16] [11]. Overcoming this limitation requires atomic-level insight into reaction mechanisms under operating conditions.
In situ Synchrotron Radiation Fourier Transform Infrared (SR-FTIR) spectroscopy has emerged as a powerful technique to directly observe the dynamic evolution of these key intermediates, providing the experimental evidence needed to design catalysts that break these scaling relationships [63]. The ultra-high brilliance of the synchrotron light source, which is 2-3 orders of magnitude brighter than conventional thermal sources, enables researchers to capture the weak and transient signals of reactive species at the electrochemical interface with superior signal-to-noise ratio (SNR) and micro-zone resolution [63]. This direct observation is crucial for validating novel catalytic mechanisms, such as the dual-site mechanism, that circumvent the traditional scaling limitations imposed by single-site catalysis [16].
Core Principles and Advantages of SR-FTIR Spectroscopy
SR-FTIR spectroscopy excels in identifying molecular structure changes by detecting vibrations of chemical bonds and functional groups, typically in the mid-infrared region (600-4000 cm⁻¹) [63]. When applied to electrocatalytic systems, it allows for the real-time monitoring of reaction intermediates adsorbed on catalyst surfaces during the reaction process.
The key advantages of SR-FTIR over conventional FTIR for studying catalytic intermediates include:
- High Brilliance: The synchrotron light source provides a photon flux that is 100 to 1000 times brighter than conventional globars, enabling high-quality data acquisition even at microscopic sampling areas [63].
- Enhanced Sensitivity: The high brilliance directly translates to a superior SNR, which is critical for detecting the low concentrations of short-lived reactive intermediates at electrode surfaces [63].
- Micro-Scale Resolution: The ability to focus on micro-zone areas (down to several micrometers) allows for the investigation of specific active sites or heterogeneous catalyst surfaces [63].
- Single-Reflection Mode Feasibility: The intense light source often makes multiple internal reflections unnecessary. Operating in single-reflection mode reduces experimental complexity and potential artifacts, providing more straightforward interpretation of interfacial phenomena [63].
The acquisition of in-situ SR-FTIR spectra on electrochemical interfaces is most effectively performed using Attenuated Total Reflection (ATR) configurations, particularly in a single-reflection mode. In this setup, the IR beam irradiates the sample at a high incident angle (optimally between 83° and 88°), penetrating only a short distance (typically ~0.5-2 µm) into the sample and generating an evanescent wave that probes the interface with minimal bulk solution interference [63].
Troubleshooting Guide: Common SR-FTIR Experimental Challenges and Solutions
Even with the advanced capabilities of SR-FTIR, researchers often encounter practical challenges that can compromise data quality. The table below summarizes common issues, their potential impact on your research, and recommended solutions.
Table 1: Common SR-FTIR Experimental Challenges and Solutions
| Problem Category | Specific Symptom | Potential Impact on Data | Recommended Solution |
|---|---|---|---|
| Instrument & Environment | Noisy spectra, strange peaks | False spectral features, inaccurate intermediate identification | Ensure instrument is on a vibration-free bench; isolate from pumps/lab activity [64]. |
| Unstable baseline, peak shifts | Inaccurate quantification of intermediate concentrations | Allow instrument sufficient warm-up time; maintain stable temperature and humidity [65]. | |
| Accessory & Setup | Negative absorbance peaks | Inability to distinguish sample signal from artifact | Clean ATR crystal thoroughly and collect a fresh background spectrum [64] [66]. |
| Distorted or saturated peaks | Loss of spectral information, incorrect functional group analysis | For diffuse reflection measurements, process data in Kubelka-Munk (K-M) units instead of absorbance [64] [66]. | |
| Sample Preparation | Spectral interference at ~3400 cm⁻¹ & ~2300 cm⁻¹ | Obscured signals from O-H, N-H, or C≡O bonds | Purge instrument and sample chamber with dry air or inert gas to remove atmospheric H₂O and CO₂ [65]. |
| Weak or distorted signals from solid samples | Poor quality spectra, missing key intermediate signals | Grind solid samples finely and uniformly; ensure even distribution in KBr pellets [65]. | |
| Spectral features change over time | Misinterpretation of reaction pathway | For volatile liquid samples, use sealed cells or perform rapid data collection to prevent evaporation [65]. | |
| Data Interpretation | Overlapping absorption bands | Difficulty assigning peaks to specific intermediates | Use spectral deconvolution techniques; consult reference databases for fingerprint regions [65]. |
| Unidentified peaks in fingerprint region | Confusion between intermediates and contaminants | Always run control experiments; be aware of combination bands and overtones from the catalyst itself [65]. |
Frequently Asked Questions (FAQs)
Q1: Why is my SR-FTIR signal for reactive intermediates still weak even after using a synchrotron source? Weak signals can originate from several factors. The most common is suboptimal alignment of the in-situ electrochemical cell, which reduces the effective light throughput. Ensure the cell's IR window (e.g., ZnSe) is clean and correctly aligned. Secondly, low surface concentration of the catalyst or intermediates will inherently yield a weak signal. Optimizing catalyst loading on the electrode and focusing the SR beam on the most active spot is crucial. Finally, excessive interference from the electrolyte can be a problem. Using a thin-layer cell configuration in ATR mode helps minimize the absorption from the bulk solution, thereby enhancing the surface sensitivity [63].
Q2: How can I distinguish between a genuine reaction intermediate and a surface contaminant in my spectrum? Genuine reaction intermediates are typically potential-dependent. Their signal intensity should grow, diminish, or shift as you change the applied electrode potential. Contaminants, on the other hand, usually display constant signals. Running control experiments is essential: perform identical procedures without the reactant (e.g., without CO₂ for CO2RR studies) or on an inert electrode substrate to establish a baseline and identify peaks originating from the electrolyte, cell components, or accidental contamination [63] [65].
Q3: What is the single most important step to ensure a high-quality in-situ SR-FTIR experiment? The most critical step is meticulous and frequent background collection. The background spectrum accounts for the contributions of the cell, atmosphere, and electrode substrate. Any change in these conditions between collecting the background and sample spectra will introduce artifacts. Always collect a new background spectrum after cleaning the ATR crystal, purging the system, and at the exact same electrode potential (preferably at the rest potential) where you intend to start your reaction. A dirty crystal during background collection is a primary source of negative peaks and distorted baselines [64] [66].
Q4: Our research aims to break scaling relationships by designing dual-site catalysts. Can SR-FTIR provide evidence for this? Yes, absolutely. SR-FTIR is uniquely positioned to provide experimental evidence for dual-site mechanisms. For instance, in the oxygen reduction reaction (ORR), the conventional scaling relationship between OOH and *OH intermediates on single-site catalysts can be broken if O₂ is cleaved via a M1–O–O–M2 bridge. SR-FTIR can directly detect the formation of this key Pt–O–O–Fe transition state and, crucially, confirm the *absence of the *OOH intermediate, thereby providing direct proof of an alternative reaction pathway that circumvents the scaling relationship [16].
Essential Experimental Protocols for Key Measurements
Protocol for In Situ SR-FTIR Monitoring of ORR Intermediates
This protocol is adapted from studies investigating Pt-Fe dual-atom catalysts to break ORR scaling relationships [16].
- Catalyst Ink Preparation: Disperse 5 mg of catalyst powder (e.g., Pt = N₂ = Fe on carbon) in a solution containing 1 mL of isopropanol and 50 µL of 5% Nafion solution. Sonicate for at least 60 minutes to form a homogeneous ink.
- Working Electrode Preparation: Pipette a precise volume (e.g., 10-20 µL) of the catalyst ink onto a polished and cleaned glassy carbon electrode. Allow the solvent to evaporate slowly at room temperature, resulting in a uniform catalyst film with a typical loading of 0.2-0.5 mg/cm².
- In Situ Electrochemical Cell Assembly: Assemble a three-electrode electrochemical cell with an ATR configuration. The working electrode is deposited on the ATR crystal (e.g., ZnSe), a Pt wire serves as the counter electrode, and a reversible hydrogen electrode (RHE) is used as the reference. Use 0.1 M HClO₄ or KOH as the electrolyte, purged thoroughly with high-purity N₂.
- SR-FTIR Data Collection:
- First, collect a background single-beam spectrum at the open circuit potential under N₂ atmosphere.
- Switch the electrolyte saturation to O₂ and hold the electrode at a specific potential.
- Collect interferograms at a resolution of 4-8 cm⁻¹ for a sufficient number of scans (typically 64-256) to achieve a good SNR.
- Repeat the measurement at progressively increasing potentials in a stepwise manner (e.g., from 1.0 V to 0.2 V vs. RHE).
- The final spectra are presented as relative absorbance, calculated as ΔR/R = (R - R₀)/R₀, where R is the single-beam spectrum at the sample potential and R₀ is the background spectrum.
Protocol for Differentiating Surface vs. Bulk Chemistry with ATR-FTIR
This protocol is critical for ensuring you are probing the catalytically active surface and not misled by bulk or migrated additives [64] [66].
- Surface Analysis: Place the "as-received" catalyst pellet or film directly on the ATR crystal and apply consistent pressure. Collect the IR spectrum.
- Bulk Analysis: Carefully remove the sample. Use a clean blade to cut the pellet or scrape the surface of the film to expose a fresh, interior surface.
- Repeat Measurement: Place this freshly exposed surface onto the ATR crystal and collect a new spectrum under identical instrument settings.
- Data Comparison: Compare the two spectra. Differences in the C-H stretch region (~2800-3000 cm⁻¹) or the fingerprint region (e.g., changes in peak ratios around 1100 cm⁻¹) indicate that surface chemistry (e.g., migrated plasticizers, surface oxidation) is different from the bulk material. The spectrum from the fresh interior is considered more representative of the true catalyst composition [66].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Materials for In Situ SR-FTIR Experiments
| Item Name | Function/Application | Key Considerations |
|---|---|---|
| ATR Crystals (ZnSe, Ge, Diamond) | Internal Reflection Element (IRE) that enables surface-sensitive measurement. | ZnSe offers a good balance of performance and cost; Diamond is robust but more expensive; Germanium provides high refractive index for shallow penetration depth [63]. |
| Potassium Bromide (KBr) | Matrix for preparing solid pellets of powder catalysts. | Must be stored in a desiccator and handled in low-humidity environments to prevent absorption of atmospheric water, which creates spectral interference [65]. |
| Nafion Perfluorinated Resin | Binder for preparing catalyst inks; proton conductor in fuel cell research. | Use diluted solutions (e.g., 0.5-5%); excessive amounts can create thick films that crack upon drying and hinder mass transport. |
| High-Purity Inert Gases (N₂, Ar) | Purging the instrument and electrolyte to remove interfering gases (O₂, CO₂). | Essential for eliminating spectral interference from atmospheric CO₂ (~2350 cm⁻¹) and water vapor [65]. |
| Deuterated Solvents (e.g., D₂O) | Solvent for electrolyte preparation to reduce IR absorption in the O-H stretch region. | Shifts the strong O-H stretching and bending vibrations to lower wavenumbers, freeing up the ~2500-3500 cm⁻¹ and ~1640 cm⁻¹ regions for analysis. |
| Thin-Layer Electrochemical Cell | In-situ cell that minimizes the path length of the IR beam through the electrolyte. | Critical for reducing the strong absorption of water and enabling the detection of weak signals from adsorbed intermediates [63]. |
Visualizing Workflows and Signaling Pathways
SR-FTIR Experimental Workflow for Intermediate Detection
Diagram Title: SR-FTIR Experimental Workflow
Dual-Site Mechanism for Breaking ORR Scaling Relationship
Diagram Title: ORR Pathways Comparing Single and Dual Site Mechanisms
Frequently Asked Questions
Q1: What do the Tafel slope values indicate about the reaction mechanism in oxygen evolution reaction (OER)? A change in the Tafel slope, as observed in materials like the dynamically constructed e-NiMoO4, indicates a shift in the reaction mechanism. For example, a high Tafel slope (around 120 mV dec⁻¹) is consistent with the Volmer step (water dissociation) being the rate-determining step. A lower Tafel slope (around 40 mV dec⁻¹) suggests the rate-determining step has shifted to a chemical recombination step (Tafel step), indicating optimized hydrogen adsorption energy and faster surface kinetics [67].
Q2: Why is my catalyst's performance degrading rapidly during electrolysis? Rapid degradation often stems from catalyst instability under operational conditions. A key strategy to mitigate this is to dynamically construct a dense epitaxial layer on the catalyst surface. This layer acts as a protective barrier, preventing the leaching of active components (such as molybdenum) and enhancing the material's durability, enabling stable operation for over 1400 hours at high current densities [67].
Q3: How can I balance specific surface area (SSA) and charge transfer in metal oxide OER catalysts? Machine learning analysis has identified a non-monotonic relationship between SSA and the Tafel slope. An optimal SSA window of 100–200 m² g⁻¹ is recommended. Within this range, the catalyst balances sufficient active site exposure with manageable interfacial impedance, leading to improved charge transfer kinetics and a lower Tafel slope [68].
Q4: What are the key factors to consider when benchmarking homogeneous electrocatalysts? When benchmarking catalysts, two main factors of merit should be addressed:
- Intrinsic Catalytic Performance: Compare "catalytic Tafel plots," which relate the turnover frequency (TOF) to the overpotential, providing a cell-independent performance metric.
- Catalyst Deactivation: Introduce the limiting turnover number as a key metric to quantify the effect of catalyst degradation during electrolysis [69].
Performance Benchmarking Data
Table 1: Performance Benchmarks for Representative Electrocatalysts
| Catalyst Material | Reaction | Overpotential (mV) @ specific current | Tafel Slope (mV dec⁻¹) | Stability / Limiting Turnover Number |
|---|---|---|---|---|
| 3D-MN NiCo₂O₄ [68] | OER | 304 @ 10 mA cm⁻² | Low (specific value not stated) | Remarkable durability |
| e-NiMoO₄ [67] | HER | 32 @ 10 mA cm⁻²; 170 @ 100 mA cm⁻²; 251 @ 200 mA cm⁻² | 45.7 | >1400 h @ 0.45 A cm⁻² |
| NiMoO₄ (control sample) [67] | HER | 238 @ 10 mA cm⁻² | 125.1 | Not specified |
Table 2: Key Reagent Solutions for Electrocatalyst Synthesis and Testing
| Research Reagent | Function / Explanation |
|---|---|
| Three-dimensional nitrogen-doped carbon networks [68] | Sacrificial template for creating 3D mesoporous structures in metal oxides, enabling high specific surface area. |
| KOH electrolyte with nickel chloride & sodium citrate [67] | Electrolyte and precursor system for cathodic electrochemical synthesis; chelating agent controls growth of epitaxial hydroxide layers. |
| Nickel Molybdate (NiMoO₄) precursor [67] | Platform material for dynamically constructing an epitaxial catalytic layer to enhance stability and performance. |
Experimental Protocols
Protocol 1: Data-Driven Design and Synthesis of a 3D Mesoporous NiCo₂O₄ OER Catalyst
This protocol outlines a machine-learning-guided synthesis for high-performance metal oxide catalysts [68].
- Database Construction: Systematically compile a database from literature containing catalyst composition, elemental types, structural morphology (e.g., Specific Surface Area), and electrochemical performance metrics (e.g., Tafel slope).
- Machine Learning Analysis: Employ ML models, such as SHapley Additive exPlanations (SHAP), to identify and prioritize key factors influencing the target property (e.g., Tafel slope). The analysis revealed SSA as a critical, non-monotonically influencing parameter.
- Catalyst Synthesis (Guided by ML):
- Template Preparation: Utilize a 3D nitrogen-doped carbon network as a sacrificial template.
- Impregnation: Adsorb metal ions (Ni²⁺, Co²⁺) onto the carbon template.
- Calcination: Perform a one-step annealing process. This step transforms the metal ions into the oxide phase (NiCo₂O₄) while simultaneously removing the carbon template. The confined ripening induced by the ultrathin carbon sheets assists in the assembly of nanoparticles into a 3D interconnected network.
- Characterization: Confirm the formation of a uniform, nanosized 3D mesoporous network (3D-MN) structure with a high SSA (optimally between 100-200 m² g⁻¹).
- Electrochemical Testing: Evaluate OER performance, confirming a low overpotential and a low Tafel slope over a wide current density range.
Protocol 2: Dynamic Construction of an Epitaxial Layer for Stable Alkaline HER
This protocol describes an interface engineering strategy to enhance both activity and durability [67].
- Substrate Synthesis: Synthesize NiMoO₄ precursor microrods via a hydrothermal method to establish a robust 3D substrate.
- Electrochemical Synthesis for Epitaxial Layer:
- Prepare a cathodic electrochemical synthesis bath containing KOH electrolyte, nickel chloride (additional Ni source), and sodium citrate (chelating agent).
- Use the NiMoO₄ precursor as the working electrode.
- Apply a cathodic potential to induce the growth of a dense Ni(OH)₂ nanodendrite layer epitaxially on the surface of the NiMoO₄ microrods. Optimize the potential, duration, and other electrochemical parameters.
- Material Characterization: Use SEM, STEM, XPS, and XAS (XANES/EXAFS) to confirm the epitaxial relationship between the NiMoO₄ core and the Ni(OH)₂ shell, the lower oxidation state of Ni in the layer, and the integrity of the Mo-containing framework.
- Interface and Performance Evaluation:
- Measure HER activity, noting the overpotential at 10, 100, and 200 mA cm⁻² and the Tafel slope.
- The optimized e-NiMoO₄ should show a significantly reduced Tafel slope, indicating an accelerated reaction mechanism.
- Test stability at high current densities (e.g., 0.45 A cm⁻²) in an industrial alkaline electrolyzer to demonstrate long-term durability (>1000 hours).
The Scientist's Toolkit: Essential Concepts & Workflows
Electrocatalyst Performance Optimization Workflow
The following diagram illustrates the interconnected strategies for designing high-performance electrocatalysts, bridging material design, interface engineering, and performance validation.
The transition from highly controlled model systems, like single-atom catalysts, to complex practical devices such as fuel cells and electrolyzers introduces significant operational challenges. While model systems offer nearly 100% atom utilization and exceptional activity in laboratory settings, their uniform active sites often limit performance in practical chemical reactions involving multiple intermediates [6]. This limitation arises from scaling relationships—fundamental thermodynamic constraints that inextricably link the binding energies of different reaction intermediates, making it impossible to optimize the energy landscape for every step in a complex catalytic cycle [70].
In practical devices, researchers must navigate these inherent scaling relationships while simultaneously addressing multifaceted operational issues including pump cavitation, gas management, sensor calibration, and maintenance protocols. This technical support center provides targeted guidance for overcoming these challenges, offering specific troubleshooting methodologies and maintenance protocols to help researchers maintain catalytic efficiency when transitioning from model systems to functional devices.
Fundamental Scaling Relationships in Catalysis
The Molecular Scaling Relationship Framework
Molecular scaling relationships quantitatively correlate the kinetic and thermodynamic parameters of electrocatalytic reactions. For the Oxygen Reduction Reaction (ORR), which serves as the cathode reaction in most fuel cells, these relationships connect the maximum turnover frequency (TOFmax) with the effective overpotential (ηeff) through linear correlations in the form:
log(TOFmax) = m(ηeff) + C [70]
These relationships reveal that catalytic efficiency depends not only on catalyst identity but also on reaction conditions including catalyst reduction potential and the pKa of the acid buffer. For iron-porphyrin-catalyzed ORR, the log(TOFmax) responds differently to changes in ηeff originating from different sources—18.5 decades in TOFmax/V in ηeff when resulting from different catalyst reduction potentials versus only 5.1 decades when arising from varying the buffer pKa [70]. This indicates that multiple scaling relationships exist for complex catalytic systems, and understanding these dependencies is crucial for optimizing practical devices.
Integrative Catalytic Pairs: Overcoming Scaling Limitations
Integrative catalytic pairs (ICPs) represent a promising strategy for overcoming the limitations imposed by scaling relationships in practical devices. Unlike single-atom catalysts with uniform active sites, ICPs feature spatially adjacent, electronically coupled dual active sites that function cooperatively yet independently [6]. This architecture provides functional differentiation within a small catalytic ensemble, enabling concerted multi-intermediate reactions that bypass the traditional scaling relationship constraints. ICPs have demonstrated enhanced activity and selectivity in complex reactions including nitrate reduction, CO2 conversion, and hydrogenation reactions [6], making them particularly valuable for practical fuel cell and electrolyzer applications.
Fuel Cell Troubleshooting Guide
External Fuel Pump Cavitation
Problem: Fuel pump cavitation reduces fuel delivery and can damage pump components.
Solution:
- Installation Position: Install the external fuel pump at or below the bottom of the fuel cell to create a gravity assist in the fuel line [71].
- Line Length: Keep the external fuel pickup/outlet line between the fuel cell and pump as short as possible to reduce the distance the pump must pull fuel [71].
External Vent Line Issues
Problem: Improperly plumbed vent lines cause vacuums and fuel surge.
Solution:
- Vertical Routing: Plumb the external vent line vertically off the vent fitting with no downward sags [71].
- Surge Prevention: Only run the vent line downward after it meets or exceeds the tallest point in the fill system to prevent fuel surge [71].
Programmable Sending Unit Calibration
Problem: Incorrect fuel level readings due to improper sender calibration.
Solution:
- Wiring Verification: Ensure proper connections: POS to 12V positive, NEG to 12V negative (ground), SEND to gauge Signal Input Terminal [71].
- Ohm Range Matching: Verify the ohm range specified on the sender matches the gauge requirements [71].
- Calibration Timing: Follow the exact calibration sequence with precise timing (2 seconds for empty, 5 seconds for full) [71].
Fuel Cell Performance Degradation
Problem: Gradual decrease in power output over time.
Solution:
- Normal Degradation: Recognize that steady decrease in output power is normal over time [72].
- Refurbishment Decision: Consider refurbishment when power output no longer meets application requirements [72].
Table: Fuel Cell Sending Unit Symptoms and Causes
| Symptom | Possible Causes |
|---|---|
| Needle does not move at all | Positive and Ground wires switched; No power; Broken wire; No ground; Ignition switch off [71] |
| Needle pegs FULL | Mismatched ohm range between sender and gauge; 12V positive connected to SEND terminal [71] |
| Needle pegs EMPTY | Mismatched ohm range; Sending unit probe tube in contact with water [71] |
| Needle does not travel full scale | Mismatched ohm range; Calibration required [71] |
Electrolyzer Troubleshooting Guide
Electrolyte Preparation and Management
Problem: Incorrect electrolyte concentration causing system errors.
Solution:
- KOH Concentration: For 90% pure caustic potash, use 1.5% concentration (7.5g KOH per 500mL water) [73].
- Gradual Mixing: Add KOH gradually; if excess causes undissolved material, add proportional water to reach 1.5% concentration [73].
- Proper Filling: Use the single screw cap on top of the electrolyzer; create suction at HHO outlet if electrolyte doesn't flow [73].
Electrolyzer Error Codes and Diagnostics
Problem: System generating specific error codes requiring diagnosis.
Solution:
- Flowchart Guidance: Consult manufacturer-specific troubleshooting flowcharts for error codes (e.g., FP01, FP03, WP_04 series) [74].
- Real-time Alarms: Rely on integrated temperature and pressure sensors with automatic shutdown systems for safety [73].
Efficiency and Operational Issues
Problem: Suboptimal electrolyzer efficiency and performance.
Solution:
- Regular Monitoring: Continuously monitor voltage, current, and gas purity parameters [73].
- Sensor Calibration: Regularly calibrate sensors and control systems to maintain accuracy [73].
- Fluid Management: Ensure electrolyte and cooling fluids are at correct levels with proper circulation [73].
Table: Hydrogen Electrolyzer Operational Parameters
| Parameter | Specification | Notes |
|---|---|---|
| Small Unit Energy Consumption | 0.29 kW/hr | Produces 150 liters of HHO per hour [73] |
| Water Consumption | ~1 liter every 10 hours | For models with water reservoirs [73] |
| Electrolyte Concentration | 1.5% KOH | For 90% pure caustic potash [73] |
| Efficiency | 1700% | With heat exchanger system [73] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Essential Research Reagents and Materials for Fuel Cell and Electrolyzer Research
| Item | Function | Application Notes |
|---|---|---|
| Caustic Potash (KOH) | Electrolyte | Provides potassium and hydroxyl ions; enhances water conductivity; prevents component corrosion [73] |
| Ultra-pure Methanol | Fuel | For methanol fuel cells; impurities cause irreversible damage [72] |
| Hydrogen Fuel | Reactant | 99.999% pure (5 nines) required to prevent catalyst contamination [72] |
| Proton Exchange Membrane | Solid electrolyte | Allows proton transport while separating electrodes; critical for PEM fuel cells and electrolyzers |
| Iron-Porphyrin Catalysts | ORR catalyst | Molecular catalyst for oxygen reduction; subject to scaling relationships [70] |
| Acid/Base Buffers | pH Control | Essential for maintaining equilibrium potentials; use 1:1 acid/conjugate base ratios [70] |
Experimental Protocols for Device Optimization
Determining Effective Overpotential in Electrolytic Systems
Purpose: To accurately determine ηeff for any electrocatalytic system, enabling direct comparison of catalytic efficiency across different experimental conditions.
Methodology:
- Prepare Buffered Solution: Use 1:1 acid (HA) and conjugate base (A−) to ensure defined [HA]/[A−] ratio, preventing homoconjugation effects in organic media [70].
- Determine Ecat/2: Obtain from cyclic voltammetry as the potential at half the catalytic current, equivalent to E1/2(FeIII/FeII) for iron-porphyrin systems [70].
- Calculate Erxn: Use the Nernst equation incorporating concentrations/pressures of all species and acid pKa [70].
- Compute ηeff: Apply the formula ηeff = Erxn - Ecat/2 [70].
Application Notes:
- For ORR in organic solvents: EO2/H2O = E°O2/H2O - (2.303RT/4F)log([H2O]²[A−]⁴/PO2[HA]⁴) - (0.0592V)pKa [70]
- Standard potentials can be estimated from aqueous standards and nonaqueous standard hydrogen potentials using thermochemical cycles [70]
Electrolyte Preparation and System Filling Protocol
Purpose: To ensure optimal electrolyte concentration and proper system priming for efficient and safe electrolyzer operation.
Methodology:
- Calculate KOH Amount: For 500mL water, measure 7.5g of 90% pure KOH for 1.5% concentration [73].
- Gradual Mixing: Add KOH gradually to water while stirring; avoid exceeding recommended concentration [73].
- System Filling:
- Open single screw cap on electrolyzer top
- Pour mixed solution through funnel into 500mL reservoir
- If electrolyte doesn't flow, create slight suction at upper HHO outlet nozzle [73]
- Hose Connection: Connect 1/4" PF68 hose to upper HHO outlet nozzle after filling [73].
System Visualization Diagrams
Fuel Cell Troubleshooting Flow
Scaling Relationship Solution Path
Frequently Asked Questions (FAQs)
Q1: What maintenance is required for hydrogen electrolyzers? A: Regular maintenance includes: periodic inspection of components (fans, valves, sensors); cleaning of electrodes and membranes to prevent fouling; continuous monitoring of performance parameters (voltage, current, gas purity); regular sensor calibration; fluid management to ensure correct electrolyte levels and circulation; and software updates for control systems [73].
Q2: Can fuel cells be integrated with renewable energy sources? A: Yes, fuel cells can be effectively integrated with renewable power sources like solar or wind to create hybrid power solutions. Although fuel cells can provide primary power, integration with renewables often creates a more effective and balanced energy system [72].
Q3: What are the practical applications of hydrogen produced by electrolyzers? A: Hydrogen from electrolyzers has versatile applications across sectors: in energy, it can fuel cells for electricity generation; industries can use it as feedstock for chemical processes; in transportation, it serves as clean fuel for fuel cells; it also enables energy storage and grid balancing [75].
Q4: How do molecular scaling relationships help optimize practical devices? A: Molecular scaling relationships connect thermodynamic and kinetic parameters, revealing how turnover frequency and overpotential depend on catalyst identity and reaction conditions. These relationships enable researchers to predictably tune catalytic metrics to achieve faster rates at lower overpotentials, directly informing practical device optimization [70].
Q5: What safety features are typically included in electrolyzers? A: Modern electrolyzers incorporate multiple safety measures including safety valves, temperature and pressure sensors, automatic shutdown systems, and real-time alarm delivery through electronic monitoring systems to ensure safe operation [73].
Q6: How long does fuel cell refurbishment typically take? A: The assessment phase is typically free and occurs quickly, but if the unit requires manufacturer technician diagnosis, the complete process including diagnosis, quotation, and repair usually takes around 4 weeks [72].
Q7: Can I use alternative electrolytes in my electrolyzer? A: Although electrolysis can be achieved with other electrolytes, it is strongly recommended to use the specified caustic potash (KOH) and water mixture as other electrolytes may damage the system or void warranties [73].
Q8: What is the typical efficiency range for electrolyzer systems? A: Efficiency varies by system design. Some systems with heat exchangers can achieve efficiencies as high as 1700%, significantly exceeding typical heater or boiler efficiencies that range from 70% to 99% depending on system type [73].
Conclusion
The concerted effort to overcome catalytic scaling relationships is ushering in a new era of theory-driven catalyst design. The foundational understanding of LSRs, combined with innovative strategies such as dynamic site regulation and dual-site cooperative mechanisms, provides a robust toolkit for breaking inherent activity limits. Experimental validation through advanced operando techniques confirms that these approaches can simultaneously optimize the energetics of multiple reaction steps, a feat previously considered unattainable. Future directions must focus on translating these atomic-scale insights into stable, industrially viable catalysts, particularly for the oxygen evolution and reduction reactions critical to renewable energy technologies. The paradigm shift from static to dynamically optimized active sites promises to unlock unprecedented efficiencies in catalytic processes central to a sustainable, carbon-neutral future.
References
- 1. Breaking linear scaling relationships in oxygen evolution ... [https://www.nature.com/articles/s41467-024-55150-9]
- 2. Conservation of the Total Order of the Axial Si—Si and N → Si ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503915/]
- 3. Bond Order Conservation Strategies in Catalysis Applied to ... [https://suncat.stanford.edu/publications/bond-order-conservation-strategies-catalysis-applied-nh3-decomposition-reaction]
- 4. Scaling relationships for adsorption energies of C2 ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250911001515]
- 5. Scaling Relationships and Computational Catalyst Design [https://pubmed.ncbi.nlm.nih.gov/27088666/]
- 6. Integrative catalytic pairs driving complex chemical reactions [https://www.nature.com/articles/s41570-025-00771-x]
- 7. Challenges in unconventional catalysis [https://www.sciencedirect.com/science/article/pii/S0920586123002043]
- 8. What is Allometric Scaling & Its Role in Drug Development? [https://www.allucent.com/resources/blog/what-allometric-scaling-and-how-does-it-support-drug-development]
- 9. Volcano plots in R: easy step-by-step tutorial [https://biostatsquid.com/volcano-plots-r-tutorial/]
- 10. Is the ∗O vs. ∗OH scaling relation intercept more relevant ... [https://www.sciencedirect.com/science/article/pii/S2667109324001970]
- 11. Breaking linear scaling relationships in oxygen evolution via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11790916/]
- 12. Unifying the ORR and OER with surface oxygen and ... [https://www.nature.com/articles/s41467-024-51605-1]
- 13. Scaling Catalyst Production: Challenges & Tips For Success [https://catalysts.com/industrial-catalyst-scale-up-challenges-strategies-for-success/]
- 14. A quick guide to the assessment of key electrochemical ... [https://www.sciencedirect.com/science/article/abs/pii/S0360319921047728]
- 15. Strategies to Break the Scaling Relation toward Enhanced ... [https://www.sciencedirect.com/science/article/pii/S2590238519302267]
- 16. Regulating the scaling relationship for high catalytic ... [https://www.nature.com/articles/s41467-022-34169-w]
- 17. Operando impedance spectroscopy with combined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11868597/]
- 18. Overcoming Energy‐Scaling Barriers: Efficient Ammonia ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11881671/]
- 19. Atomic-scale self-rearrangement of hetero-metastable phases into high-density single-atom catalysts for the oxygen evolution reaction | Nature Communications [https://www.nature.com/articles/s41467-025-58163-0]
- 20. Photocatalysis Lecture 4 | Fundamentals of Overpotential [https://www.perfectlight.com.cn/technology/detail-55.html]
- 21. Scaling Relations in Oxygen Electrocatalysis and Beyond [https://chemrxiv.org/engage/chemrxiv/article-details/67ed469081d2151a02b33a98]
- 22. Breaking the linear scaling relationship in BN-supported ... [https://www.sciencedirect.com/science/article/abs/pii/S1005030224010661]
- 23. Excited-State Intramolecular Proton Transfer: A Short ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7963178/]
- 24. Engineering the regulation strategy of active sites to ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979725009865]
- 25. Accelerating O-O bond dissociation in oxygen reduction ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433225003824]
- 26. Troubleshooting of Catalytic Reactors | PPTX [https://www.slideshare.net/slideshow/troubleshooting-of-catalytic-reactors/130602843]
- 27. Troubleshooting Guide for Naphtha Catalytic Reforming Units [https://www.linkedin.com/pulse/troubleshooting-guide-naphtha-catalytic-reforming-da-silva-mba--wesdf]
- 28. Transforming photocatalytic hydrogen production efficiency [https://www.sciencedirect.com/science/article/abs/pii/S0360319925011590]
- 29. Multifunctional catalytic activity of Cu3N (001) surface [https://www.sciencedirect.com/science/article/pii/S2772571522000602]
- 30. Confinement effect induced efficient electro-catalytic ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433223015672]
- 31. Catalytic confinement effects in nanochannels: from biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9418946/]
- 32. Overcoming the reactivity-stability challenge in water ... [https://www.nature.com/articles/s41467-025-64684-5]
- 33. Proton donor/acceptor effects on electrochemical ... [https://www.sciencedirect.com/science/article/abs/pii/S2451910323001709]
- 34. Regulating the scaling relationship for high catalytic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9613657/]
- 35. How Characterization Can Identify the Root Cause [https://catalysts.com/catalyst-deactivation/]
- 36. Heterogeneous catalysis [https://en.wikipedia.org/wiki/Heterogeneous_catalysis]
- 37. Identifying the Elusive Sites of Tyrosyl Radicals in Cytochrome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4063442/]
- 38. Forwarding Molecular Design of Heterogeneous Catalysts [https://pmc.ncbi.nlm.nih.gov/articles/PMC6161065/]
- 39. Catalyst Characterization Techniques [https://www.hidenanalytical.com/blog/catalyst-characterization-techniques/]
- 40. Heterogeneous Catalyst - an overview [https://www.sciencedirect.com/topics/chemistry/heterogeneous-catalyst]
- 41. Reactor Troubleshooting and Solutions [https://www.jinzongmachinery.com/a-news-reactor-troubleshooting-and-solutions.html]
- 42. Troubleshooting Process Control Problems [https://www.aiche.org/resources/publications/cep/2020/may/troubleshooting-process-control-problems]
- 43. Optimization of plasma-thermal system for non-oxidative ... [https://www.sciencedirect.com/science/article/abs/pii/S0306261925001412]
- 44. Synergy of dual-atom catalysts deviated from the scaling ... [https://www.nature.com/articles/s41467-023-40177-1]
- 45. Advances in dual-site mechanisms for designing high ... [https://www.sciencedirect.com/science/article/pii/S2667141725000333]
- 46. Breaking scaling relations with inverse catalysts [https://arxiv.org/html/2504.16493v1]
- 47. Mutually exclusive experiments [https://support.optimizely.com/hc/en-us/articles/39074082140045-Mutually-exclusive-experiments]
- 48. Sequential Design of Experiments (SDOE) - FOQUS [https://foqus.readthedocs.io/en/3.2.0/chapt_sdoe/]
- 49. Deactivation and regeneration of the supported bimetallic ... [https://www.sciencedirect.com/science/article/abs/pii/S0926337313002816]
- 50. Deactivation mechanism and regeneration effect of bi- ... [https://www.sciencedirect.com/science/article/abs/pii/S001623612102785X]
- 51. Deactivation Mechanism and Anti- ... [https://www.mdpi.com/2073-4433/14/5/770]
- 52. How to prevent selective leaching (dealloying) in metals? [https://armoloy.com/faq/how-to-prevent-selective-leaching/]
- 53. Deactivation mechanisms and regeneration of a bimetallic ... [https://www.sciencedirect.com/science/article/abs/pii/S0167299199804923]
- 54. Reversible and Irreversible Deactivation of Supported Bimetallic ... [https://link.springer.com/article/10.1023/A:1010409230898]
- 55. Leaching techniques to remove metals and potentially ... [https://www.sciencedirect.com/science/article/abs/pii/S0043135411005148]
- 56. Xray Absorption Near Edge Spectroscopy- XANES [https://www.cei.washington.edu/education/science-of-solar/xray-absorption-near-edge-spectroscopy-xanes/]
- 57. Best practices for in-situ and operando techniques within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11911412/]
- 58. Tracking dynamic structural changes in catalysis by rapid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8415324/]
- 59. Improving the quality of XAFS data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6038603/]
- 60. 1.3 Reaction Mechanisms and Rate-Determining Steps [https://fiveable.me/physical-chemistry-ii/unit-1/reaction-mechanisms-rate-determining-steps/study-guide/S4KDuLxtHLPc8pX0]
- 61. 4.6b: Reaction Mechanisms [https://chem.libretexts.org/Courses/Mount_Royal_University/Chem_1202/Unit_4%3A_Chemical_Kinetics/4.6b%3A_Reaction_Mechanisms]
- 62. Remediation of heavy metals contaminated soil by enhanced ... [https://arabjchem.org/remediation-of-heavy-metals-contaminated-soil-by-enhanced-electrokinetic-technology-a-review/]
- 63. In-Situ Synchrotron Radiation Infrared Spectroscopic ... [http://www.ccspublishing.org.cn/article/doi/10.14102/j.cnki.0254-5861.2022-0083]
- 64. FT-IR Troubleshooting Guide: How to Solve Common ... [https://www.spectroscopyonline.com/view/ftir-troubleshooting-guide-how-to-solve-common-problems-in-infrared-spectroscopy]
- 65. Common Problems and Precautions in the Operation of ... [https://www.drawellanalytical.com/common-problems-and-precautions-in-the-operation-of-ftir-spectrophotometer/]
- 66. Common Problems with FT-IR Instruments and How to ... [https://www.spectroscopyonline.com/view/common-problems-with-ft-ir-instruments-and-how-to-avoid-them]
- 67. Dynamic construction of a durable epitaxial catalytic layer ... [https://www.nature.com/articles/s41467-025-63361-x]
- 68. Data-driven design of metal oxide electrocatalysts with low ... [https://www.sciencedirect.com/science/article/abs/pii/S0925838825025253]
- 69. of homogeneous electrocatalysts: Benchmarking ... overpotential [https://pubmed.ncbi.nlm.nih.gov/25757058/]
- 70. Developing Scaling Relationships for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7351079/]
- 71. Troubleshooting Guide [https://fuelsafe.com/pages/troubleshooting-guide]
- 72. Fuel Cell Systems FAQs | Answers to Common Questions [https://www.fuelcellsystems.co.uk/faqs]
- 73. FAQ - Efena [https://efenahydrogen.com/faq-page/]
- 74. Troubleshooting: EL 2.1 [https://handbook.enapter.com/troubleshooting/el21/]
- 75. Hydrogen Electrolyzers: Five Frequently Asked Questions [https://www.plugpower.com/blog/hydrogen-electrolyzers-five-frequently-asked-questions/]