AI vs. Traditional Catalyst Development: A 2025 Comparison of Speed, Cost, and Innovation in Drug Discovery
This article provides a comprehensive analysis for researchers and drug development professionals on the paradigm shift from traditional, trial-and-error catalyst development to AI-driven approaches.
AI vs. Traditional Catalyst Development: A 2025 Comparison of Speed, Cost, and Innovation in Drug Discovery
Abstract
This article provides a comprehensive analysis for researchers and drug development professionals on the paradigm shift from traditional, trial-and-error catalyst development to AI-driven approaches. We explore the foundational principles of both methods, detail the application of machine learning and automated robotics in modern catalyst design, and address key challenges like data quality and model interpretability. Through a comparative validation of real-world case studies and performance metrics, we demonstrate how AI is accelerating timelines, reducing R&D costs, and enabling the discovery of novel catalytic materials, ultimately shaping the future of biomedical research.
The Catalyst Development Paradigm Shift: From Intuition to Algorithm
Traditional catalyst development has long been characterized by a research paradigm deeply rooted in iterative, human-led experimentation. This methodology relies almost exclusively on the specialized knowledge and intuition of experienced researchers, who manually design experiments based on established chemical principles and historical data. The process is fundamentally guided by empirical relationships and linear free energy relationships (LFERs)—such as the Brønsted catalysis law, Hammett equation, and Taft equation—which provide simplified, quantitative insights into structure-activity relationships based on limited, curated datasets [1]. Before the advent of sophisticated computational planning tools, chemists depended heavily on database search engines like Reaxys and SciFinder to retrieve published reaction information, a process limited to previously recorded transformations and unable to guide the discovery of novel catalysts or unreported synthetic routes [1].
The core scientific challenge within this traditional research paradigm lies in the immense complexity and high dimensionality of the search space, which encompasses virtually limitless variables related to catalyst composition, structure, reactants, and synthesis conditions [2]. Other significant limitations include the general lack of data standardization and the inherently lengthy research cycles, which not only consume substantial manpower and material resources but also introduce considerable uncertainty into research outcomes [2]. This article provides a detailed comparison of this established approach against emerging AI-driven methodologies, examining their respective experimental protocols, performance data, and practical implications for research efficiency.
Core Methodologies and Workflows
The Traditional Experimental Workflow
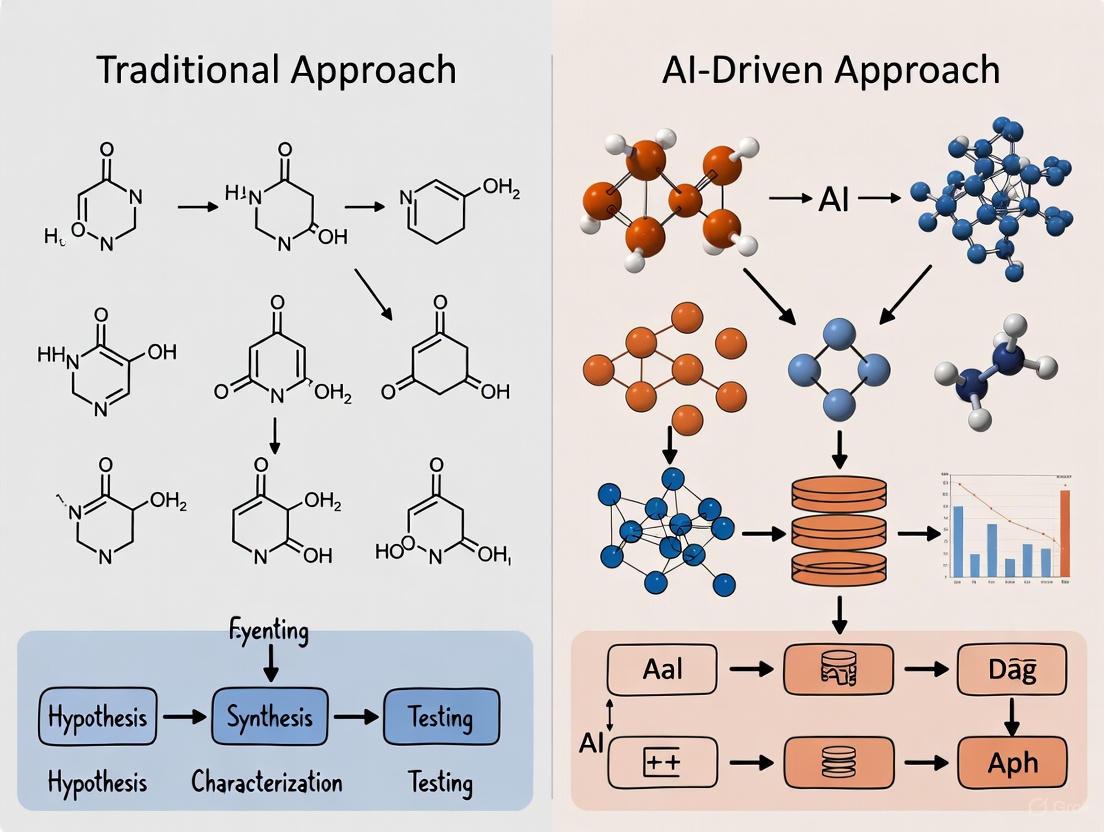
The conventional pathway for developing catalysts is a sequential, labor-intensive process that progresses through distinct, manually-executed stages. Figure 1 illustrates this iterative cycle, which is heavily dependent on human intervention at every step.
Diagram Title: Traditional Catalyst Development Workflow
As shown in Figure 1, the process begins with Hypothesis Formulation, where researchers design catalyst candidates based on prior literature, chemical intuition, and known descriptor-property relationships [2] [1]. This is followed by Manual Catalyst Design, focusing on optimizing composition and structure to achieve target activity and stability.
The Trial-and-Error Synthesis stage involves manually testing factors like precursor selection, temperature, time, solvent, and atmospheric environment. These factors significantly influence the final product's composition, structure, and morphology, and their interplay makes controlled synthesis particularly challenging [2]. Subsequently, Performance Evaluation assesses catalytic activity, selectivity, and stability, while Data Analysis and Interpretation relies on researcher expertise to derive insights. The loop repeats based on these findings, creating a Lengthy Feedback Loop that typically spans months or even years [2] [3].
The AI-Driven Experimental Workflow
In contrast, AI-driven catalyst development employs a closed-loop, autonomous workflow that integrates artificial intelligence, robotics, and real-time data analysis. Figure 2 outlines this accelerated, data-rich process, which minimizes human intervention.
Diagram Title: AI-Driven Catalyst Development Workflow
As shown in Figure 2, the AI-driven process starts with AI Model Prediction, where machine learning models virtually screen millions of potential compositions and structures by identifying patterns in large datasets to predict promising candidates [2] [3]. This enables Automated High-Throughput Synthesis using robotic systems and self-driving laboratories to synthesize shortlisted candidates [2] [4]. The Automated Characterization and Performance Testing stage employs integrated analytical instruments and real-time monitoring (e.g., NMR) for high-throughput evaluation [5]. Finally, Real-Time Data Processing and Machine Learning Analysis automatically processes results to refine AI models, creating a Rapid Feedback Loop that can take just hours or days [5] [4].
Performance Comparison: Quantitative Data Analysis
Key Performance Metrics
The fundamental differences between traditional and AI-driven methodologies become strikingly apparent when comparing their performance across key metrics. Table 1 summarizes quantitative data from direct comparisons and representative case studies.
Table 1: Performance Comparison of Traditional vs. AI-Driven Catalyst Development
| Performance Metric | Traditional Approach | AI-Driven Approach | Experimental Context |
|---|---|---|---|
| Development Timeline | Years to decades [3] | Months to years [3] | Discovery of new catalyst materials [2] |
| Experimental Throughput | 30 tests/day (manual) [6] | 30 tests/day (automated) [6] | High-throughput catalyst testing [6] |
| Number of Candidates Screened | ~1,000 candidates [3] | >19,000 candidates [6] | Virtual screening for HER catalysts [6] |
| Success Rate in Synthesis | Not explicitly stated | <25% of targets matched [6] | Mixed-metal catalyst synthesis [6] |
| Resource Optimization | High (manual resource use) | 9.3x improvement in power density per dollar [4] | Fuel cell catalyst discovery [4] |
| Heat Duty Reduction | Baseline (100%) | 38% of baseline [7] | Catalyst-aided CO₂ desorption [7] |
Detailed Experimental Protocols
Traditional Protocol: Catalyst-Aided CO₂ Desorption
This established protocol for testing solid acid catalysts in CO₂ desorption exemplifies the traditional, sequential experimental approach [7].
- Objective: To evaluate the performance of solid acid catalysts (γ-Al₂O₃ and HZSM-5) in reducing the energy required for CO₂ desorption from loaded amine solvents in a pilot plant setting.
- Materials & Setup:
- Solvent System: Blended 5M monoethanolamine (MEA) and 2M methyl diethanolamine (MDEA), compared to benchmark 5M MEA.
- Catalysts: Industrial solid acid catalysts γ-Al₂O₃ (Lewis acid) and HZSM-5 (Bronsted acid).
- Reactor: Packed-bed desorber column operated at 1 atm.
- Setup: Modified desorption process with a heat exchanger replacing the traditional reboiler, enabling operation at temperatures below 100°C.
- Procedure:
- The rich amine solvent (pre-loaded with CO₂) is fed into the desorber column at a constant flow rate (60 mL/min).
- The solvent flows through the packed bed containing the solid acid catalyst.
- Desorption is carried out at atmospheric pressure (1 bar) and temperatures below 100°C.
- The lean CO₂ loading of the solvent exiting the desorber is measured.
- The CO₂ production rate is quantified and used to calculate the heat duty (energy requirement).
- Key Outcome: The combined action of the catalysts with the blended solvent decreased the heat duty from the baseline of 100% to 38%, significantly reducing energy consumption [7].
AI-Driven Protocol: Self-Driving Laboratory for Fuel Cell Catalysts
This protocol describes the AI-driven, closed-loop workflow used by the CRESt platform to discover advanced fuel cell catalysts [4].
- Objective: To autonomously discover a multielement catalyst for a direct formate fuel cell that achieves high power density while minimizing precious metal content.
- Materials & Setup:
- AI Platform: CRESt (Copilot for Real-world Experimental Scientists) integrating multimodal data and robotic equipment.
- Robotic Equipment: Liquid-handling robot, carbothermal shock synthesizer, automated electrochemical workstation.
- Characterization Tools: Automated electron microscopy, X-ray diffraction.
- Precursors: Up to 20 different precursor molecules and substrates included in the search space.
- Procedure:
- AI Prediction: The system searches scientific literature and uses active learning to propose promising catalyst recipes.
- Automated Synthesis: Robotic systems execute the synthesis of proposed catalysts.
- Automated Testing & Characterization: The performance of each catalyst is tested in a fuel cell setup, with parallel characterization of its structure.
- Real-Time Analysis: Data from experiments are fed back into the AI models, which use Bayesian optimization to refine the search space and suggest the next set of experiments.
- Iteration: Steps 1-4 are repeated autonomously over hundreds of cycles.
- Key Outcome: After exploring over 900 chemistries and conducting 3,500 tests, CRESt discovered an 8-element catalyst that achieved a 9.3-fold improvement in power density per dollar compared to pure palladium [4].
The Scientist's Toolkit: Essential Research Reagents and Materials
The experimental approaches, both traditional and AI-driven, rely on a specific set of chemical reagents, catalysts, and instrumentation. Table 2 details these key research solutions and their functions.
Table 2: Key Research Reagent Solutions and Materials
| Item Name | Type/Classification | Primary Function in Experimentation |
|---|---|---|
| Monoethanolamine (MEA) | Solvent (Primary Amine) | Benchmark absorbent for CO₂ in post-combustion capture; forms carbamate ions with CO₂ [7]. |
| Methyl Diethanolamine (MDEA) | Solvent (Tertiary Amine) | Used in blended amine solvents to promote bicarbonate ion formation, improving overall capture and desorption performance [7]. |
| γ-Al₂O₃ | Catalyst (Lewis Acid Solid Acid) | Facilitates desorption by replacing the role of bicarbonate in the reaction mechanism, lowering energy requirements [7]. |
| HZSM-5 | Catalyst (Bronsted Acid Solid Acid) | Provides protons to aid in the breakdown of carbamate during CO₂ desorption [7]. |
| Palladium (Pd) | Catalyst (Precious Metal) | Benchmark precious metal catalyst for hydrogenation and fuel cell reactions; expensive but highly active [4] [6]. |
| VSP-P1 Printer | Instrumentation (Synthesizer) | Automated device that vaporizes metal rods to create nanoparticles of desired composition for high-throughput catalyst synthesis [6]. |
| Periodic Open-Cell Structures (POCS) | Reactor Component (Structured Reactor) | 3D-printed architectures (e.g., Gyroids) that provide superior heat and mass transfer compared to conventional packed beds [5]. |
| Benzene-1,4-dicarboxylate | Ligand (Linker in MOFs) | Common organic linker used in the synthesis of Metal-Organic Frameworks (MOFs) for catalytic applications [2]. |
The comparative analysis clearly demonstrates a paradigm shift in catalyst development. The traditional approach, while built on a deep foundation of chemical expertise and historical data, is inherently limited by its sequential nature, low throughput, and extensive reliance on manual effort. This results in prolonged development cycles spanning years or decades and a constrained ability to explore complex, multi-element chemical spaces [2] [3].
In contrast, the AI-driven approach represents a transformative advancement. By integrating machine learning, robotics, and high-throughput experimentation, it enables the rapid screening of thousands to millions of candidates, the identification of non-intuitive catalyst compositions, and a drastic reduction in development time and cost [4] [6]. The integration of AI is not merely an incremental improvement but a fundamental re-engineering of the research workflow, paving the way for accelerated discovery of advanced catalysts critical to addressing pressing challenges in energy and sustainability.
The field of catalysis research is undergoing a profound transformation, moving from traditional trial-and-error approaches to data-driven, artificial intelligence (AI)-powered methodologies. Catalysts, which accelerate chemical reactions without being consumed, are fundamental to modern industry, playing critical roles in energy production, pharmaceutical development, and environmental protection [2] [3]. Historically, catalyst development has been a time-consuming and resource-intensive process, often relying on empirical observations, intuition, and sequential experimentation that can span years [2] [8]. This traditional paradigm faces significant challenges in navigating the highly complex and multidimensional search spaces of catalyst composition, structure, and synthesis conditions [2].
The integration of machine learning (ML) is sharply transforming this research paradigm, offering unique advantages in tackling highly complex issues across every aspect of catalyst synthesis [2]. AI provides powerful new capabilities for identifying descriptors for catalyst screening, processing massive computational data, fitting potential energy surfaces with exceptional accuracy, and uncovering mathematical laws for chemical and physical interpretability [2]. This article provides a comprehensive comparison between traditional and AI-driven catalyst development approaches, examining their respective methodologies, performance metrics, and implications for research efficiency and catalyst performance.
Comparative Analysis: Traditional vs. AI-Driven Catalyst Development
Table 1: Fundamental Characteristics of Traditional and AI-Driven Catalyst Development Approaches
| Aspect | Traditional Approach | AI-Driven Approach |
|---|---|---|
| Core Methodology | Trial-and-error experimentation, empirical observations, sequential testing [2] [8] | Data-driven prediction, virtual screening, algorithmic optimization [2] [3] |
| Development Timeline | Years to decades [3] | Months to years [3] |
| Primary Resource Investment | Laboratory equipment, reagents, human labor [2] | Computational infrastructure, data acquisition, specialized expertise [2] [9] |
| Data Handling | Limited, often inconsistent datasets; reliance on published literature and isolated experiments [2] [10] | Large-scale, standardized datasets; high-throughput experimentation generating thousands of data points [2] [10] |
| Key Limitations | High cost, lengthy cycles, cognitive biases, difficulty optimizing multiple parameters simultaneously [2] [10] | Data quality dependencies, model generalizability challenges, "black box" interpretability issues [3] [9] |
| Optimization Capability | Limited to few variables at a time; local optimization [10] | High-dimensional parameter space navigation; global optimization [2] [10] |
Table 2: Performance and Outcome Comparison
| Performance Metric | Traditional Approach | AI-Driven Approach |
|---|---|---|
| Experimental Efficiency | Low: Testing 1,000 catalysts requires synthesizing all 1,000 candidates [3] | High: AI narrows field to 10 most promising candidates from 1,000 possibilities [3] |
| Success Rate Prediction | Limited to empirical trends and theoretical models with simplified systems [10] | Enhanced: 92% accuracy demonstrated in knowledge extraction tasks [9] |
| Multi-Objective Optimization | Challenging: Difficulty balancing activity, selectivity, stability simultaneously [3] | Promising: ML models can predict trade-offs between multiple performance descriptors [2] [10] |
| Discovery of Novel Materials | Serendipitous or incremental improvements based on existing knowledge [8] | Systematic exploration of chemical space; prediction of entirely new catalytic systems [11] [9] |
| Scalability | Limited by manual processes [2] | High: Enabled by automated high-throughput systems [2] |
| Knowledge Extraction | Manual literature review; limited integration of disparate studies [9] | Automated: Natural language processing of scientific literature [11] [9] |
Experimental Protocols in AI-Driven Catalyst Research
The Catal-GPT Framework for Catalyst Design
A pioneering example of AI implementation in catalysis is the Catal-GPT framework, which employs a large language model (LLM) specifically fine-tuned for catalyst design [9]. The experimental protocol involves:
Data Collection and Curation: A specialized web crawler navigates academic databases to extract chemical data from scientific abstracts, which is then cleaned and encoded into a model-readable format [11] [9]. When conflicting parameters appear for the same catalytic system, priority is given to preparation parameters from authoritative publications with the highest reported frequency [9].
Model Architecture and Training: The system uses the open-source qwen2:7b LLM, deployed locally with a specialized database on the oxidative coupling of methane (OCM) reaction. The architecture is modular, comprising data storage, foundation model, agent, and feedback learning modules [9].
Knowledge Extraction and Validation: The model undergoes task evaluations for knowledge extraction and research assistance. In testing, it achieved 92% accuracy in knowledge extraction and could propose complete catalyst preparation processes, including required chemical reagents and detailed synthesis parameters [9].
Iterative Optimization: The system incorporates feedback from experimental results or industrial applications to continuously refine its recommendation strategy, creating a dynamic learning loop [9].
Machine Learning with Experimental Descriptors
For experimental catalysis research, ML models utilize descriptors encompassing catalyst composition, synthesis variables, and reaction conditions [10]. The protocol typically involves:
Descriptor Selection: Input features may include catalyst composition (presence of metals, functional groups), synthesis parameters (calcination temperature, precursor selection), and reaction conditions (temperature, pressure) [10].
Model Training: Using tree-based algorithms (decision trees, random forests, XGBoost) for classification tasks and regression algorithms (linear regression, gradient boost decision tree) for predicting continuous variables like faradaic efficiency [10].
Feature Importance Analysis: Determining the relative significance of experimental factors through techniques like descriptor importance analysis, which examines prominence and frequency during the decision process of tree-based models [10].
Iterative Design: Using ML predictions to guide subsequent experimental rounds, progressively narrowing the search space and refining catalyst formulations [10].
AI-Driven Catalyst Discovery Workflow: This diagram illustrates the iterative, data-driven cycle of AI-assisted catalyst development, from initial data collection through model training, prediction, experimental validation, and continuous refinement.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Computational Tools in AI-Driven Catalysis
| Tool/Reagent | Function/Application | Role in AI-Driven Research |
|---|---|---|
| High-Throughput Synthesis Systems (e.g., AI-EDISON, Fast-Cat) [2] | Automated preparation of catalyst libraries | Generates large, consistent datasets essential for training robust ML models [2] |
| Specialized LLMs (e.g., Catal-GPT, ChemCrow, ChemLLM) [9] | Natural language processing of scientific literature | Extracts knowledge from vast research corpus; suggests catalyst formulations [11] [9] |
| Descriptor Libraries [10] | Mathematical representations of catalyst properties | Encodes complex chemical information into machine-readable features for model input [10] |
| Web Crawlers & Data Extraction Tools [11] | Automated mining of scientific databases | Builds comprehensive datasets from published literature for AI training [11] |
| Robotic AI Chemists [2] | Autonomous synthesis and testing | Enables closed-loop experimentation with minimal human intervention [2] |
| Transfer Learning Frameworks [11] | Application of knowledge across chemical domains | Enhances predictive capabilities when experimental data is limited [11] |
The comparison between traditional and AI-driven catalyst development approaches reveals a complementary relationship rather than a simple replacement scenario. While AI methodologies offer unprecedented speed in screening catalyst candidates and ability to navigate complex parameter spaces, traditional experimental expertise remains crucial for validating predictions, interpreting results, and guiding model development [2] [8]. The most promising path forward involves the integration of physical knowledge and mechanistic understanding with data-driven AI approaches, creating a synergistic workflow that leverages the strengths of both paradigms [10].
The future of catalysis research lies in increasingly autonomous systems, with AI not only suggesting catalyst compositions but also planning and executing synthetic routes, performing characterizations, and iterating based on experimental outcomes [2]. As these technologies mature, they promise to significantly accelerate the development of catalysts for critical applications including renewable energy, environmental protection, and sustainable chemical production [3] [8].
The field of catalyst development is undergoing a profound transformation, moving from a tradition steeped in empirical methods to one increasingly guided by data-driven prediction. For decades, the discovery and optimization of catalysts have relied heavily on trial-and-error experimentation—a resource-intensive process constrained by human intuition, time, and cost. This approach, while responsible for many critical advances, is inherently limited when navigating the vast complexity of chemical and biological catalyst spaces. In contrast, a new paradigm is emerging, one that integrates high-throughput experimentation, large-scale data generation, and artificial intelligence (AI) to predict catalytic behavior and design novel systems rationally. This guide objectively compares these two core philosophies, examining their fundamental principles, methodologies, performance, and practical implications for researchers and scientists in drug development and related fields.
Fundamental Principles: A Philosophical Divide
The traditional and data-driven approaches are founded on fundamentally different philosophies for navigating scientific discovery.
The Trial-and-Error Philosophy: The traditional approach is largely empirical and iterative. It relies on the chemist's intuition and prior knowledge to formulate initial hypotheses about promising catalysts or reaction conditions. Experiments are then designed and executed sequentially. The outcome of each experiment informs the next, creating a slow, cyclical process of refinement. This method is inherently local in its exploration; researchers typically make small, incremental changes to known systems (e.g., slightly modifying a ligand or a reaction temperature) rather than venturing into entirely uncharted chemical territory. The process is often described as resource-intensive, with success heavily dependent on researcher experience and serendipity [12] [13].
The Predictive, Data-Driven Philosophy: This modern approach treats catalyst discovery as a global optimization problem within a vast, multidimensional space. Its core principle is that patterns embedded in large, high-quality datasets can be used to build models that accurately predict catalytic performance. Instead of relying solely on chemical intuition, this method uses machine learning (ML) to identify complex, non-linear relationships between catalyst features (descriptors) and their functional outcomes (e.g., activity, selectivity). The goal is to shift the experimental burden from blind screening to targeted validation of computationally prioritized candidates, fundamentally accelerating the discovery process [12] [14] [13].
Methodological Comparison: Experimental Protocols in Practice
The practical implementation of these two philosophies differs significantly in workflow, techniques, and tools.
Traditional Trial-and-Error Workflow
The classical protocol is linear and iterative [13]:
- Hypothesis Formulation: Based on literature review and expert knowledge, a chemist identifies a potential catalyst or a set of reaction conditions for a target transformation.
- Sequential Experimentation: The lead candidate is synthesized and tested in the lab. This process is often low-throughput, with one reaction or a small batch of reactions run at a time.
- Analysis and Interpretation: Results (e.g., yield, conversion, selectivity) are analyzed.
- Iterative Refinement: The chemist uses the results to make an educated guess for the next experiment, perhaps by tuning a single variable (e.g., ligand concentration, solvent). This process loops back to step 2.
- Optimization and Scale-Up: Once a promising candidate is identified, further rounds of experimentation optimize its performance before moving to larger-scale synthesis.
This workflow is visualized in the following diagram:
Data-Driven Design Workflow
The AI-driven approach creates a closed-loop, cyclical system that integrates computation and experimentation [12] [14]:
- Data Acquisition and Curation: A large, high-quality dataset is assembled. This can be from historical records, high-throughput experiments, or computational simulations. For example, in biocatalysis, this might involve screening hundreds of enzymes against a library of substrates to create a dataset like BioCatSet1 [12].
- Descriptor Engineering and Feature Selection: Raw data (e.g., molecular structures, protein sequences) is converted into numerical descriptors that a machine can process. Feature selection techniques may be used to identify the most relevant parameters.
- Model Training and Validation: A machine learning algorithm (e.g., Gradient Boosted Decision Trees, Random Forest) is trained on the dataset to learn the mapping between input descriptors and output performance. The model is rigorously validated on unseen data.
- Prediction and Candidate Prioritization: The trained model is used to screen a vast virtual library of potential catalysts, predicting their performance and generating a ranked list of the most promising candidates.
- Targeted Experimental Validation: Only the top-predicted candidates are synthesized and tested in the lab, drastically reducing experimental workload.
- Data Feedback and Model Retraining: Results from validation experiments are fed back into the dataset, retraining and improving the model for the next iteration in a continuous improvement cycle.
This workflow is visualized in the following diagram:
Quantitative Performance Comparison
The following tables summarize experimental data and performance metrics from case studies that directly or indirectly compare the efficiency and outcomes of the two approaches.
Table 1: Comparative Efficiency in Biocatalyst Discovery (CATNIP Case Study) [12]
| Performance Metric | Traditional High-Throughput Screening | AI-Guided Prediction (CATNIP Model) |
|---|---|---|
| Initial Experimental Scale | 314 enzymes × 111 substrates (~34,854 reactions) | N/A (Leverages prior data) |
| Hit Identification Rate | Baseline (Random) | 7x higher than random screening |
| Key Experimental Step | Test all combinations | Validate only top 10 model-predicted enzymes |
| Validation Success Rate | N/A (Discovery method) | 70-80% (7 out of 10 predicted enzymes were active) |
| Exploration Nature | Broad but shallow "fishing expedition" | Targeted "spear fishing" in chemical space |
Table 2: Performance of AI-Driven Workflows in Catalyst Design (Selected Examples)
| Application / Model | Key Performance Metric | Traditional/DFT Method | AI/Data-Driven Method |
|---|---|---|---|
| SurFF Surface Model [15] | Computational Efficiency for Surface Energy | Density Functional Theory (DFT) | ~100,000x faster than DFT |
| CaTS Framework [15] | Transition State Search Efficiency | Standard DFT Calculation | ~10,000x faster than DFT |
| CO₂ to Methanol SAC Screening [15] | Catalyst Screening Throughput | Low (DFT bottleneck) | Screening of 3,000+ candidates; discovery of new high-performance SACs |
| CATNIP (Enzyme → Substrate) [12] | Discovery of Novel Reactions | Limited to known enzyme functions | Successful prediction and validation of multiple unprecedented biocatalytic reactions |
The Scientist's Toolkit: Essential Research Reagents & Materials
This section details key reagents, software, and materials essential for implementing the data-driven design workflow, as featured in the cited research.
Table 3: Key Reagent Solutions for Data-Driven Catalyst Development
| Item Name | Function/Description | Example from Research |
|---|---|---|
| Enzyme Library (aKGLib1) [12] | A diverse collection of biological catalysts for high-throughput experimental screening to generate training data. | A library of 314 NHI enzymes with average sequence identity of 13.7%, ensuring high diversity. |
| Substrate Library [12] | A collection of diverse small molecules used to probe the catalytic activity and specificity of catalysts. | A library of >100 compounds, including chemical building blocks, natural products, and drug molecules. |
| Functional Monomers [16] | Building blocks for data-driven polymer design, selected to represent classes of amino acids. | Six monomers representing hydrophobic, nucleophilic, acidic, cationic, amide, and aromatic classes. |
| Sequence Similarity Network (SSN) [12] | A bioinformatics tool to visualize and analyze sequence relationships, used for selecting diverse enzyme candidates. | Used to select the 314 enzymes for aKGLib1 from a pool of 265,632 sequences. |
| Machine Learning Model (e.g., GBM) [12] | The algorithmic core that learns from data to make predictions; GBM was used in CATNIP. | Gradient Boosted Decision Tree model for linking chemical and protein sequence spaces. |
| MORFEUS Software [12] | Computational chemistry tool for calculating molecular "fingerprints" or descriptors for small molecules. | Used to compute a set of 21 parameters for each substrate as input for the ML model. |
The contrast between traditional trial-and-error and predictive, data-driven design marks a pivotal shift in scientific methodology for catalyst development. The empirical approach, while foundational, is constrained by its sequential nature, high resource costs, and limited capacity to explore vast chemical spaces. In contrast, the data-driven paradigm, powered by AI and high-throughput experimentation, offers a powerful strategy for global exploration and predictive accuracy. It does not seek to eliminate experimentation but to make it profoundly more efficient and insightful by guiding it with intelligent prediction.
For researchers and drug development professionals, the implication is clear: integrating data-driven approaches into the R&D pipeline can dramatically accelerate discovery timelines, reduce costs associated with failed experiments, and unlock novel catalytic functions that might remain hidden under traditional methodologies. The future of catalyst design lies in the continued refinement of this closed-loop paradigm—"experiment-data-AI"—where each cycle of prediction and validation generates deeper, more actionable scientific understanding [12] [14].
For researchers and scientists in drug development and chemical synthesis, the traditional approach to catalyst design has long been a critical bottleneck. The conventional trial-and-error methodology, reliant on empirical observations and sequential experimentation, consumes substantial resources while delivering incremental progress. This paradigm is now being fundamentally transformed by artificial intelligence, which offers a new framework for catalyst discovery and optimization. As the catalyst market continues to expand—projected to reach USD 76.7 billion by 2033—the imperative for more efficient development approaches becomes increasingly urgent across research institutions and industrial laboratories [17]. This comparison guide examines the quantitative and methodological distinctions between traditional and AI-driven catalyst development, providing experimental data and protocols to inform research direction and resource allocation.
Quantitative Comparison: Traditional vs. AI-Driven Catalyst Development
Table 1: Performance Metrics Comparison Between Traditional and AI-Driven Catalyst Development
| Performance Metric | Traditional Approach | AI-Driven Approach | Experimental Basis |
|---|---|---|---|
| Discovery Timeline | Years to decades | Months to weeks | AI systems explored 900+ chemistries in 3 months [4] |
| Experimental Throughput | 10-100 samples manually | 3,500+ tests automated | Robotic platforms enabled 3,500 electrochemical tests [4] |
| Resource Consumption | High (reagents, labor) | Reduced by 90%+ | AI targets 10 most promising from 1,000 candidates [3] |
| Success Rate Optimization | Incremental improvements | 9.3x performance improvement | Record power density in fuel cells with reduced precious metals [4] |
| Data Utilization | Limited, experiential | Multimodal integration | Combines literature, experimental data, and characterization [4] |
Table 2: Economic and Operational Impact Analysis
| Impact Area | Traditional Approach | AI-Driven Approach | Supporting Data |
|---|---|---|---|
| Development Cost | High (extensive lab work) | Significant reduction | AI reduces experiments, lowering reagent and labor costs [3] |
| Return on Investment | Long-term, uncertain | $3.70 return per $1 invested | Demonstrated in generative AI applications [18] |
| Personnel Requirements | Large teams | Smaller, specialized teams | 32% of organizations expect AI-related workforce changes [19] |
| Scale-up Transition | High failure rate | Improved prediction | Digital twins simulate industrial conditions [3] |
| Environmental Impact | Higher waste generation | Greener processes | Enables lower temperature/pressure reactions [3] |
Experimental Protocols and Methodologies
Protocol 1: AI-Driven High-Throughput Catalyst Screening
Objective: Rapid identification of novel catalyst formulations with target properties using integrated AI-robotic systems.
Materials and Equipment:
- Liquid-handling robot for precise precursor dispensing
- Carbothermal shock system for rapid synthesis
- Automated electrochemical workstation for performance testing
- Scanning electron microscope with automated image analysis
- X-ray diffraction equipment for structural characterization
- Computational infrastructure for machine learning models
Methodology:
- Goal Definition: Researchers input target catalytic properties (e.g., activity, selectivity) through natural language interface [4]
- Literature Mining: AI system scans scientific literature and databases to identify potential elements and molecular configurations [4]
- Knowledge Embedding: Creates multidimensional representations of catalyst recipes based on prior knowledge [4]
- Design Space Reduction: Applies principal component analysis to identify most promising experimental regions [4]
- Robotic Synthesis: Automated systems prepare catalyst candidates across identified compositional space [2]
- High-Throughput Characterization: Parallel testing of catalytic performance, structure, and morphology [20]
- Active Learning Loop: Experimental results feed back into AI models to refine subsequent experimental designs [4]
Data Analysis:
- Machine learning models correlate compositional and structural features with performance metrics
- Computer vision algorithms analyze characterization data (SEM, XRD) for quality assessment
- Bayesian optimization identifies promising regions for subsequent experimentation
Protocol 2: Traditional Catalyst Optimization
Objective: Systematic improvement of catalyst formulations through sequential experimentation.
Materials and Equipment:
- Standard laboratory glassware and reactors
- Manual catalyst synthesis equipment
- Analytical instruments (GC-MS, HPLC)
- Performance testing apparatus
Methodology:
- Hypothesis Formation: Based on researcher experience and literature review
- Bench-Scale Synthesis: Manual preparation of catalyst candidates
- Performance Testing: Sequential evaluation of activity, selectivity, and stability
- Characterization: Structural analysis to understand performance characteristics
- Iterative Refinement: Modification of synthesis parameters based on results
- Scale-up Studies: Transition from laboratory to pilot scale
Data Analysis:
- Statistical analysis of experimental results
- Empirical correlation of synthesis parameters with performance
- Expert interpretation of characterization data
Workflow Visualization: Traditional vs. AI-Driven Approaches
AI-Driven vs Traditional Catalyst Development Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for AI-Driven Catalyst Development
| Reagent/Material | Function in Research | Application Examples | AI Integration |
|---|---|---|---|
| Precious Metals (Pd, Pt) | Active catalytic sites for key reactions | Fuel cells, emission control, pharmaceutical synthesis | ML models optimize loading and distribution [2] |
| Base Metals (Fe, Ni, Cu) | Cost-effective catalytic elements | Ammonia production, bulk chemicals | AI identifies optimal coordination environments [4] |
| Zeolites & MOFs | High-surface-area catalyst supports | Petrochemical refining, selective oxidation | ML guides morphology engineering [2] |
| Metal Oxides & Nitrides | Stable catalytic materials for harsh conditions | Water splitting, environmental catalysis | AI predicts stability and activity [14] |
| Enzyme Biocatalysts | Selective biological catalysts | Pharmaceutical intermediates, fine chemicals | AI models protein structures for enhanced activity [21] |
| Bimetallic Nanomaterials | Enhanced activity and selectivity | Fuel cells, specialized chemical synthesis | AI optimizes elemental combinations [2] |
| Robotic Synthesis Platforms | Automated catalyst preparation | High-throughput experimentation | Executes AI-designed experiments [4] |
| Multimodal AI Systems | Integrated data analysis and prediction | Catalyst design across applications | Processes literature, experimental data, characterization [4] |
The comparative analysis presented in this guide demonstrates a fundamental shift in catalyst development paradigms. AI-driven approaches consistently outperform traditional methods across critical metrics: reducing discovery timelines from years to months, improving resource efficiency through targeted experimentation, and enabling more predictive scale-up transitions. The integration of multimodal AI systems with robotic experimentation platforms represents a particularly significant advancement, creating closed-loop discovery systems that continuously refine their experimental strategies based on real-time results [4]. For research organizations and drug development professionals, the adoption of AI-enhanced catalyst development is transitioning from competitive advantage to operational necessity. This transition requires not only technological investment but also methodological adaptation—embracing workflow redesign, data-driven decision making, and interdisciplinary collaboration between domain experts and data scientists. As AI capabilities continue to advance, particularly in areas of interpretability and physical insight integration, the efficiency gains in catalyst development are likely to accelerate, potentially transforming not only how catalysts are designed but also what chemical transformations become economically viable.
Inside the AI-Driven Lab: Machine Learning, Automation, and Real-World Workflows
The field of catalyst development is undergoing a profound transformation, moving from traditional, intuition-guided experimental methods to artificial intelligence (AI)-driven, data-centric approaches. Traditional catalyst discovery has historically relied on trial-and-error experimentation, a process that is often slow, resource-intensive, and limited in its ability to explore vast compositional and structural spaces [2]. In stark contrast, AI-driven methodologies leverage machine learning (ML), generative models, and high-throughput computational screening to predict catalyst composition, structure, and activity with unprecedented speed and accuracy [2] [22] [23]. This paradigm shift is not merely an incremental improvement but a fundamental change in research methodology, enabling the discovery of novel, high-performance catalysts for applications in energy, sustainability, and pharmaceuticals at an accelerated pace. This guide provides a comparative analysis of these two approaches, supported by experimental data and detailed methodologies.
Comparative Analysis: Traditional vs. AI-Driven Catalyst Development
The following table summarizes the core differences between the traditional and AI-driven catalyst development paradigms.
Table 1: Core Differences Between Traditional and AI-Driven Catalyst Development
| Aspect | Traditional Approach | AI-Driven Approach |
|---|---|---|
| Core Methodology | Trial-and-error experimentation, literature guidance, and linear hypothesis testing [2]. | Data-driven prediction, high-throughput virtual screening, and generative design [2] [22]. |
| Exploration Speed | Months to years for a single discovery cycle; limited by manual synthesis and testing [2]. | Days to weeks; capable of screening millions of candidates computationally [23] [3]. |
| Data Utilization | Relies on limited, localized datasets; knowledge often remains within individual research groups [24]. | Leverages large, shared databases (e.g., >400,000 data points) and learns from every cycle in a closed-loop system [24]. |
| Key Capabilities | Density Functional Theory (DFT) calculations, standard characterization techniques [2]. | Machine learning interatomic potentials (MLIPs), generative adversarial networks (GANs), variational autoencoders (VAEs), and transformer models [2] [22]. |
| Primary Limitations | High cost, low throughput, inability to efficiently navigate vast design spaces, and lengthy research cycles [2] [3]. | Dependency on data quality and quantity, challenges in model interpretability ("black box" issue), and integration with experimental validation [2] [3]. |
Performance and Outcome Comparison
Quantitative data from recent studies highlights the dramatic performance advantages of AI-driven catalyst design.
Table 2: Quantitative Performance Comparison of Catalyst Discovery Methods
| Metric | Traditional & Computational | AI-Driven Method | Result |
|---|---|---|---|
| Materials Discovered | ~28,000 materials discovered via computational approaches over a decade [23]. | 2.2 million new crystals predicted by GNoME; 380,000 classified as stable [23]. | ~80x increase in discovered stable materials. |
| Discovery Rate | ~50% accuracy in stability prediction from earlier models [23]. | Over 80% prediction accuracy achieved by GNoME via active learning [23]. | ~60% relative improvement in prediction accuracy. |
| Efficiency Gain | Standard computational screening methods [25]. | New ML method predicts material structure with five times the efficiency of the previous standard [25]. | 5x more efficient in structure prediction. |
| Layered Compounds | ~1,000 known graphene-like layered compounds [23]. | GNoME discovered 52,000 new layered compounds [23]. | 52x more potential candidates for electronics. |
Experimental Protocols in AI-Driven Catalyst Design
The Closed-Loop Workflow of the Digital Catalysis Platform (DigCat)
The DigCat platform exemplifies a comprehensive, cloud-based AI workflow for autonomous catalyst design [24].
- AI-Driven Proposal: A researcher submits a query (e.g., "design a new catalyst"). The platform's AI agent, integrating large language models (LLMs) and material databases, generates initial candidate compositions and structures [24].
- Stability and Cost Screening: Candidates undergo automated stability analysis, including surface Pourbaix diagram and aqueous stability assessment, to filter for practical applicability [24].
- Activity Prediction: Machine learning regression models predict adsorption energies and activities. Candidates are screened using traditional thermodynamic volcano plot models and further evaluated with machine learning force fields [24].
- Performance Validation: Selected candidates are input into pH-dependent microkinetic models for reactions like oxygen reduction (ORR) or CO2 reduction (CO2RR). These models account for electric field-pH coupling, kinetic barriers, and solvation effects [24].
- Automated Synthesis and Testing: The most promising candidates are automatically synthesized by robotic platforms (e.g., at partner institutions like Tohoku University). High-throughput experimental devices collect performance and characterization data [24].
- Closed-Loop Feedback: Experimental results are fed back into the DigCat platform. The AI agent uses this data to update its machine learning and physical models, improving the accuracy of subsequent design cycles [24].
Generative Design of Surfaces and Adsorbates
Generative models create novel catalyst structures by learning from existing data.
- Data Collection: A dataset of crystal structures, surfaces, and/or adsorption geometries is compiled, either from public databases (e.g., Materials Project) or via custom global structure searches [22] [26].
- Model Training: A generative model, such as a Crystal Diffusion Variational Autoencoder (CDVAE) or a diffusion model, is trained on the dataset. The model learns the underlying probability distribution of stable atomic arrangements [22].
- Conditional Generation: The trained model generates new structures. This can be guided by desired properties (e.g., low adsorption energy for a specific intermediate) by conditioning the generation process on these properties [22].
- Stability and Activity Assessment: The generated structures are evaluated for stability using DFT calculations or MLIPs. Their catalytic activity is predicted via surrogate models or detailed microkinetic modeling [22].
- Experimental Validation: Promising generated candidates are synthesized and tested. For example, a study using a CDVAE and optimization algorithm generated 250,000 candidates, leading to the synthesis and validation of five new alloy catalysts for CO2 reduction, two with Faradaic efficiencies near 90% .
Workflow Visualization
The following diagram illustrates the integrated, closed-loop nature of a modern AI-driven catalyst discovery platform.
The Scientist's Toolkit: Essential Research Reagents and Solutions
This table details key computational and experimental tools essential for conducting research in AI-driven catalyst design.
Table 3: Essential Research Reagents and Solutions for AI-Driven Catalyst Design
| Tool / Solution | Function | Application in Workflow |
|---|---|---|
| Density Functional Theory (DFT) | Provides high-fidelity calculations of electronic structure, energies, and reaction barriers [22] [3]. | Generating training data for ML models; final validation of AI-predicted candidates. |
| Machine Learning Interatomic Potentials (MLIPs) | Surrogate models that provide DFT-level accuracy at a fraction of the computational cost [22]. | Accelerating molecular dynamics simulations and structure relaxation during virtual screening. |
| Generative Models (VAEs, GANs, Diffusion) | AI models that create novel molecular and crystal structures from learned data distributions [22]. | Inverse design of new catalyst compositions and surface structures with targeted properties. |
| Graph Neural Networks (GNNs) | ML architectures that operate on graph data, naturally representing atomic connectivity [23]. | Predicting material stability and functional properties directly from crystal structure (e.g., in GNoME). |
| Microkinetic Modeling Software | Simulates the detailed kinetics of catalytic reactions over a surface, accounting for all elementary steps [24]. | Predicting the overall activity and selectivity of candidate catalysts under realistic conditions. |
| Automated Synthesis Robotics | Robotic platforms that execute material synthesis protocols without human intervention [24] [2]. | High-throughput synthesis of AI-predicted catalyst candidates for experimental validation. |
| High-Throughput Characterization | Automated equipment for rapid performance testing (e.g., activity, selectivity) and structural analysis [24] [2]. | Providing rapid experimental feedback to close the AI design loop. |
The field of molecular catalysis is undergoing a profound transformation, moving from a discipline historically guided by chemist intuition and trial-and-error to one increasingly driven by data-driven artificial intelligence (AI) approaches [1]. This shift is particularly critical for navigating complex reaction conditions, where multidimensional variables—including temperature, pressure, catalyst composition, and solvent systems—interact in ways that often challenge conventional optimization strategies [14]. The comparison between traditional and AI-driven methodologies represents more than a simple technological upgrade; it constitutes a fundamental reimagining of the catalyst development workflow with substantial implications for efficiency, cost, and discovery rates across chemical industries including pharmaceutical development [27].
Traditional catalyst development has relied heavily on established principles such as linear free energy relationships (LFERs), including the Brønsted catalysis law and Hammett equation, which provided elegant but simplified structure-activity relationships based on limited datasets [1]. While these approaches have yielded significant successes over decades of research, they struggle to address the intricate interplay of factors in complex catalytic systems. In contrast, AI-driven approaches leverage machine learning (ML) to identify patterns and predict outcomes directly from high-dimensional, complex datasets, enabling researchers to explore vast chemical spaces with unprecedented efficiency and precision [1] [14]. This comparative analysis examines the performance, methodologies, and practical implementation of these competing paradigms, providing researchers with objective data to inform their experimental strategies.
Comparative Performance: Traditional vs. AI-Driven Catalyst Development
Quantitative Performance Metrics
Rigorous evaluation of both traditional and AI-driven approaches reveals significant differences in efficiency, accuracy, and resource allocation. The table below summarizes key performance indicators derived from recent research implementations:
Table 1: Performance Comparison of Traditional vs. AI-Driven Catalyst Development
| Performance Metric | Traditional Approach | AI-Driven Approach | Experimental Context |
|---|---|---|---|
| Screening Efficiency | 10-100 candidates/month [14] | 10,000+ candidates/in silico cycle [14] | High-throughput catalyst screening |
| Prediction Accuracy | ~60-70% for novel systems [1] | 85-95% for target properties [14] | Catalyst activity/selectivity prediction |
| Optimization Cycle Time | 3-6 months per development cycle [27] | 1-4 weeks per iteration [1] | Reaction condition optimization |
| Resource Utilization | High (specialized equipment, materials) [28] | Primarily computational [14] | Catalyst development cost analysis |
| Success Rate for Novel Discovery | <5% for de novo design [1] | 12-25% for validated discoveries [14] | Experimental validation of predictions |
Experimental Validation Data
The theoretical advantages of AI-driven approaches are substantiated by experimental data across diverse catalytic applications. In retrosynthetic planning, AI tools like ASKCOS and AiZynthFinder have demonstrated the capability to design viable synthetic routes for complex molecules with success rates exceeding 80% in experimental validation studies [1]. For catalyst design specifically, ML models predicting catalytic activity have achieved correlation coefficients (R²) of 0.85-0.95 with experimental validation data, significantly outperforming traditional descriptor-based models that typically achieve R² values of 0.60-0.75 [14].
In autonomous experimentation systems integrating AI with robotics, researchers have demonstrated the optimization of complex reaction conditions in hours or days—a process that traditionally required months. One documented study achieved a 15% yield improvement for a challenging catalytic transformation within 72 hours of autonomous optimization, compared to an average of 4 months using traditional sequential optimization approaches [1]. These performance differentials become particularly pronounced for systems with high-dimensional parameter spaces, where AI methods can simultaneously optimize 5-10 variables compared to the practical limit of 2-3 variables using traditional one-variable-at-a-time (OVAT) approaches [14].
Methodological Comparison: Experimental Protocols and Workflows
Traditional Catalyst Development Workflow
Traditional catalyst development follows a linear, hypothesis-driven approach grounded in chemical intuition and established principles. The typical workflow consists of the following standardized protocol:
- Literature Review and Mechanism Analysis: Researchers conduct extensive manual review of published literature on analogous catalytic systems, applying established linear free-energy relationships and mechanistic principles to formulate initial hypotheses [1].
- Catalyst Library Design: Based on periodic table trends, known structure-activity relationships, and ligand effects, a limited library of 10-50 candidate catalysts is designed, often focusing on structural analogs of known performers [28].
- Sequential Experimental Testing: Candidates are synthesized and tested sequentially using one-variable-at-a-time (OVAT) methodology, where reaction conditions (temperature, solvent, concentration) are varied individually while keeping other parameters constant [28] [29].
- Product Analysis and Characterization: Reaction products are characterized using standardized analytical techniques (GC-MS, HPLC, NMR) to determine key performance metrics (yield, conversion, selectivity) [29].
- Mechanistic Studies: For promising candidates, additional kinetic studies, isotopic labeling, and spectroscopic analysis are conducted to elucidate reaction mechanisms and deactivation pathways [28].
- Iterative Optimization: Based on results, slight structural modifications are made over multiple cycles (3-10 iterations), with each cycle requiring complete re-synthesis and re-testing [1].
This traditional workflow, while methodologically sound, inherently limits the exploration of chemical space due to practical constraints on time and resources [14].
AI-Driven Catalyst Development Workflow
AI-driven catalyst development employs an integrated, data-driven workflow that fundamentally reengineers the discovery process. The standardized protocol encompasses:
- Data Curation and Feature Engineering: Existing experimental data (literature, in-house databases) is aggregated and standardized. Molecular descriptors are computed, including electronic, structural, and topological features, with feature selection algorithms identifying the most relevant parameters [14].
- Model Selection and Training: Appropriate machine learning algorithms (random forest, neural networks, Gaussian process regression) are selected based on dataset size and complexity. The model is trained on historical data to learn complex relationships between catalyst features and performance metrics [14].
- In Silico Catalyst Screening: The trained model predicts performance for thousands of virtual candidates in the chemical space, prioritizing the most promising candidates for experimental validation [1] [14].
- Automated Experimental Validation: Robotic high-throughput systems synthesize and test top-predicted candidates (10-100 compounds) in parallel, generating standardized performance data [1].
- Active Learning Loop: Experimental results are fed back to refine the AI model, which then suggests the next round of candidates or optimal conditions, creating a continuous improvement cycle [14].
- Mechanistic Interpretation: Advanced interpretation techniques (SHAP analysis, sensitivity analysis) identify which molecular features most strongly influence performance, providing mechanistic insights [14].
This iterative, data-driven workflow enables more efficient exploration of chemical space and faster convergence on optimal solutions [1].
Workflow Visualization
The fundamental differences between these approaches are visualized in the following workflow diagrams:
Diagram 1: Traditional Catalyst Development Workflow
Diagram 2: AI-Driven Catalyst Development Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementation of both traditional and AI-driven approaches requires specific research tools and platforms. The following table details essential solutions currently employed in the field:
Table 2: Essential Research Reagent Solutions for Catalyst Development
| Tool/Category | Specific Examples | Function & Application | Compatibility |
|---|---|---|---|
| Retrosynthesis Software | ASKCOS [1], AiZynthFinder [1], Chemitica [1] | Computer-aided synthesis planning; de novo route design for catalyst precursors & target molecules | Both approaches |
| Descriptor Calculation | RDKit [1], Dragon, COMSI | Computes molecular features (electronic, topological) as inputs for QSAR models & AI algorithms | Primarily AI-driven |
| Machine Learning Platforms | Scikit-learn [14], TensorFlow [14], PyCXTM | Builds predictive models for catalyst properties & reaction outcomes | Primarily AI-driven |
| High-Throughput Experimentation | Automated liquid handlers, flow chemistry reactors [1], parallel pressure reactors | Accelerates experimental validation of AI predictions; enables rapid data generation | Primarily AI-driven |
| Catalyst Libraries | Commercial metal salt collections, ligand libraries (e.g., Solvias, Sigma-Aldrich) | Provides physical compounds for traditional screening & initial training data for AI | Both approaches |
| Data Management Systems | Electronic Lab Notebooks (ELNs), Chemical Information Systems (e.g., Reaxys, SciFinder) | Manages experimental data, literature knowledge, and historical results | Both approaches |
The comparative analysis reveals that AI-driven and traditional approaches to catalyst development offer complementary strengths rather than representing mutually exclusive alternatives. AI methodologies demonstrate clear superiority in screening efficiency, optimization speed, and handling high-dimensional parameter spaces [1] [14]. However, traditional approaches provide essential mechanistic understanding, validate AI predictions, and offer intuitive guidance that remains valuable for interpreting complex chemical phenomena [28] [1].
The emerging paradigm for optimizing complex reaction conditions involves strategic integration of both approaches, leveraging AI for rapid exploration and initial optimization while employing traditional methods for mechanistic verification and refinement of top-performing candidates [1] [14]. This hybrid model represents the most promising path forward, combining the scale and efficiency of data-driven discovery with the deep chemical insight of traditional catalysis research. As AI tools continue to evolve—particularly in areas of interpretability and integration with automated experimental platforms—this synergistic approach is poised to dramatically accelerate the development of advanced catalytic systems for pharmaceutical synthesis and other chemical applications [1] [14].
The field of chemical research is undergoing a profound transformation, shifting from traditional, labor-intensive trial-and-error approaches toward intelligent, autonomous discovery engines. Autonomous laboratories, which integrate robotic platforms with artificial intelligence (AI), are closing the traditional "predict-make-measure" discovery loop, enabling accelerated exploration of chemical space with minimal human intervention [30]. These self-driving laboratories represent the convergence of several advanced technologies: chemical science databases, large-scale AI models, automated experimental platforms, and integrated management systems that work synergistically to create a seamless research environment [30]. In catalyst development—a field historically constrained by extensive experimentation—this paradigm shift is particularly impactful, offering the potential to dramatically compress development timelines from years to weeks while optimizing for performance, cost, and sustainability [9].
This comparison guide examines the fundamental differences between traditional and AI-driven catalyst development approaches, providing researchers and drug development professionals with a comprehensive analysis of performance metrics, experimental methodologies, and the essential technological toolkit required for modern autonomous research. By objectively comparing these paradigms through quantitative data and detailed protocols, we aim to illuminate both the transformative potential and current limitations of autonomous workflow systems in chemical research.
Performance Comparison: Traditional vs. AI-Driven Catalyst Development
The quantitative advantages of AI-driven autonomous workflows become evident when comparing key performance metrics across multiple dimensions of catalyst development. The following table synthesizes experimental data from recent implementations of closed-loop systems.
Table 1: Performance Comparison of Catalyst Development Approaches
| Performance Metric | Traditional Approach | AI-Driven Autonomous Approach | Experimental Context |
|---|---|---|---|
| Development Timeline | Months to years [9] | Weeks to months [9] | Oxidative Coupling of Methane (OCM) catalyst development [9] |
| Compounds Synthesized per Candidate | Thousands [31] | Hundreds [31] | CDK7 inhibitor program [31] |
| Design Cycle Efficiency | Baseline | ~70% faster cycles [31] | Exscientia's small-molecule design platform [31] |
| Knowledge Extraction Accuracy | Manual literature review | 92% accuracy [9] | Qwen2:7b LLM on OCM catalyst data [9] |
| Success Rate | Industry standard | 10x fewer compounds synthesized [31] | AI-designed clinical candidates [31] |
| Data Utilization | Limited, unstructured | Multimodal, structured, real-time [30] | Autonomous laboratory platforms [30] |
The performance differential stems from fundamental operational distinctions. Traditional catalyst development relies heavily on researcher intuition and sequential experimentation, where each iteration requires manual intervention, data interpretation, and hypothesis generation [9]. In contrast, autonomous systems implement continuous design-make-test-analyze cycles where AI models rapidly propose new experiments based on all accumulated data, robotic platforms execute these experiments with high precision and reproducibility, and the results immediately inform subsequent cycles [30]. This closed-loop operation not only accelerates the empirical screening process but also enables more efficient exploration of complex parameter spaces through Bayesian optimization and other machine learning algorithms that strategically prioritize the most promising experimental directions [30] [9].
Experimental Protocols in Autonomous Catalyst Development
Catal-GPT Workflow for OCM Catalyst Optimization
A representative experimental protocol for AI-driven catalyst development is demonstrated by the Catal-GPT system for oxidative coupling of methane (OCM) catalysts, which employs a structured, iterative workflow [9]:
Data Curation and Knowledge Base Construction: The process begins with assembling a comprehensive dataset of OCM catalyst formulations, preparation parameters, and performance metrics extracted from scientific literature. In the Catal-GPT implementation, this involved collecting data on catalyst synthesis, characterization, and application, followed by data cleaning and encoding to fit the model's input format. Conflicting parameters for the same catalytic system were resolved by prioritizing preparation parameters from authoritative publications with the highest reported frequency [9].
Natural Language Interface and Query Processing: Researchers interact with the system through natural language queries (e.g., "Suggest a La2O3-based catalyst with high C2 selectivity"). The large language model (qwen2:7b in this case) processes these queries by extracting relevant knowledge from the structured database and generating specific formulation suggestions [9].
Hypothesis Generation and Experimental Planning: The AI model proposes complete catalyst preparation processes, including specific chemical reagents, concentrations, calcination temperatures, and other critical parameters. For example, the system might recommend a La2O3/CaO catalyst with precise molar ratios and calcination at 800°C based on optimized patterns learned from the training data [9].
Robotic Execution and Synthesis: Automated robotic platforms execute the suggested synthesis protocols. While the Catal-GPT study focused on the AI component, integrated platforms like the University of Science and Technology of China's autonomous laboratory employ robotic systems for actual catalyst preparation, handling powder processing, mixing, heating, and other synthesis steps with minimal human intervention [30].
Characterization and Performance Testing: The synthesized catalysts undergo automated characterization and testing. For OCM catalysts, this typically includes catalytic testing in continuous-flow reactors with online gas chromatography to measure methane conversion, C2+ selectivity, and yield under standardized conditions [9].
Data Integration and Model Refinement: Experimental results are fed back into the database, creating a learning loop where the AI model continuously refines its predictions based on empirical evidence. This feedback learning module allows the system to progressively improve its recommendation accuracy over multiple iterations [9].
Autonomous Workflow Orchestration
The execution of autonomous experiments relies on formal workflow models that ensure robust operation. These workflows are typically represented as state machines: F=(S,s0,T,δ)F = (S, s0, T, \delta), where SS represents possible states, s0s0 the start state, TT terminal states, and δ\delta the transition function between states based on observations [32]. Advanced systems implement dynamic scheduling where task readiness is determined by: ready(t)⟺∀p∈parents(t),status(p)=Doneready(t) \iff \forall p \in parents(t),\ status(p)=\mathrm{Done}, ensuring proper dependency management throughout the experimental sequence [32].
System Architecture and Workflow Visualization
The operational framework of an autonomous laboratory can be visualized as an integrated system where digital intelligence continuously directs physical experimentation. The following diagram illustrates the core closed-loop workflow and its key components.
Autonomous Laboratory Closed-Loop Workflow
This architecture creates a self-optimizing system where each component plays a critical role. The chemical science database serves as the foundational knowledge base, integrating multimodal data from proprietary databases, open-access platforms, and scientific literature, often structured using knowledge graphs for efficient retrieval [30]. Large-scale intelligent models, including both specialized algorithms like Bayesian optimization and genetic algorithms, and large language models like Catal-GPT, provide the cognitive engine for experimental planning and prediction [30] [9]. Automated experimental platforms physically execute the experiments through robotic systems that handle synthesis, formulation, and characterization tasks with precision and reproducibility [30]. Finally, management and decision systems orchestrate the entire workflow, dynamically allocating resources, managing experimental queues, and ensuring fault tolerance through checkpointing and automatic retries [32].
The Researcher's Toolkit: Essential Technologies and Platforms
Implementing autonomous workflows requires a sophisticated technology stack that spans computational, robotic, and data infrastructure. The following table details key solutions and their functions in enabling closed-loop research.
Table 2: Essential Research Reagent Solutions for Autonomous Chemistry
| Technology Category | Representative Platforms/Tools | Primary Function | Application in Autonomous Workflows |
|---|---|---|---|
| AI/LLM Platforms | Catal-GPT [9], ChemCrow [9], ChemLLM [9] | Catalyst formulation design, knowledge extraction, reaction prediction | Generating executable catalyst preparation methods; extracting knowledge from literature |
| Workflow Orchestration | AlabOS [32], Globus Flows [32], Emerald [32] | Experimental workflow management, resource scheduling, fault tolerance | Orchestrating complex experimental sequences with dynamic resource allocation |
| Robotic Automation | Automated robotic platforms [30], AutomationStudio [31] | High-throughput synthesis, sample processing, characterization | Physically executing chemical synthesis and analysis with minimal human intervention |
| Data Management | Chemical science databases [30], SAC-KG framework [30] | Structured data storage, knowledge graph construction, data retrieval | Organizing multimodal chemical data for AI model training and retrieval |
| Simulation & Modeling | Density functional theory (DFT) [30], Molecular dynamics [9] | Theoretical calculation, property prediction, mechanism elucidation | Providing prior knowledge and validation for AI-generated hypotheses |
| Cloud Infrastructure | Amazon Web Services (AWS) [31], Cloud offloading frameworks [32] | Scalable computing, data storage, platform integration | Hosting AI models and providing computational resources for data analysis |
This technology stack enables the implementation of increasingly sophisticated autonomous systems. For example, the integration of Catal-GPT for catalyst design with AlabOS for workflow orchestration and robotic automation platforms creates a cohesive system that can autonomously propose, execute, and optimize catalyst development campaigns [9] [32]. The emergence of cloud-native platforms like Exscientia's AWS-integrated system demonstrates how scalable infrastructure supports the substantial computational demands of these workflows, particularly when incorporating foundation models and large-scale data analysis [31].
The comparison between traditional and AI-driven catalyst development approaches reveals a fundamental shift in research methodology with profound implications for efficiency, cost, and discovery potential. Autonomous workflows consistently demonstrate superior performance across multiple metrics, particularly in accelerating development timelines, reducing material requirements, and enabling more systematic exploration of complex chemical spaces [30] [9] [31]. However, these systems face ongoing challenges including data quality requirements, model generalization limitations, and the need for specialized expertise to implement and maintain the complex technology stack [9].
The trajectory of autonomous laboratories points toward increasingly integrated and intelligent systems. Future developments will likely focus on enhancing AI models through deeper integration with physical simulations like density functional theory, establishing standardized knowledge graphs for improved data extraction, and creating more sophisticated multi-agent architectures where specialized AI modules collaborate on complex research problems [9] [32]. As these technologies mature, autonomous workflows are poised to transition from specialized implementations to mainstream research infrastructure, potentially redistributing human researcher roles from routine experimentation to higher-level strategic planning, interpretation, and innovation [30] [33]. This evolution promises to not only accelerate catalyst development but fundamentally expand the boundaries of explorable chemical space, opening new frontiers in materials science, drug discovery, and sustainable energy technologies.
The field of oncology therapeutic development stands at a transformative crossroads, marked by the convergence of traditional drug discovery methodologies with cutting-edge artificial intelligence technologies. Traditional drug discovery has long been characterized by extensive timelines averaging 10-15 years, exorbitant costs exceeding $1-2.6 billion, and dauntingly low success rates, with only 4-7% of investigational new drug applications ultimately gaining approval [34]. This inefficient paradigm has created significant bottlenecks in delivering life-saving treatments to cancer patients, particularly for those with rare or treatment-resistant malignancies. The integration of AI technologies is fundamentally reshaping this landscape, accelerating discovery timelines, improving success rates, and enabling the targeting of previously undruggable pathways [34] [35].
The underlying transformation represents a fundamental shift from "experience-driven" to "data-driven" research paradigms, mirroring similar revolutions occurring across scientific disciplines. In catalyst design, for instance, machine learning has demonstrated the capability to accelerate computational screening by factors of up to 10⁵ times compared to traditional density functional theory calculations [36]. Similarly, in oncology drug discovery, AI platforms are now compressing the initial discovery timeline from target identification to clinical candidate selection from 4-5 years to as little as 18-24 months [35]. This case study examines the quantitative performance differences between traditional and AI-driven approaches, analyzes the experimental methodologies enabling these advances, and explores the implications for the future of oncology therapeutics development.
Quantitative Comparison: Traditional vs. AI-Driven Drug Discovery
Table 1: Performance Metrics Comparison Between Traditional and AI-Driven Drug Discovery Approaches
| Performance Metric | Traditional Approach | AI-Driven Approach | Improvement Factor |
|---|---|---|---|
| Discovery Timeline | 4-5 years [35] | 18-24 months [35] | 60-70% reduction |
| Cost per Candidate | ~$1-2.6 billion [34] [37] | Significant reduction [35] | Not fully quantified |
| Phase 1 Success Rate | Industry standard: ~40-50% [38] | AI-designed molecules: 80-90% [38] | 2x improvement |
| Target Identification | Limited to known pathways | Hundreds to thousands of novel targets [37] | Order of magnitude increase |
| Molecular Screening | Months for limited libraries | Hours for billions of molecules [37] | 10⁵ times acceleration [36] |
| Clinical Trial Recruitment | Manual screening, slow enrollment | AI-matching, reduced screening time [38] | Significant efficiency gains |
Table 2: AI Platform Performance in Specific Oncology Drug Discovery Applications
| AI Platform/Technology | Application | Reported Performance | Clinical Stage |
|---|---|---|---|
| Exscientia - Centaur Chemist | OCD treatment (DSP-1181) | First AI-designed drug to reach trials [35] | Phase 1 |
| Insilico Medicine - Pharma.AI | TNIK inhibitor for IPF (ISM001-055) | Target identification to PCC in 18 months [35] | Phase IIa |
| Recursion - Phenotypic Screening | CCM disease (REC-994) | Identified novel compound for rare disease [35] | Phase II (terminated) |
| Schrödinger - Physics-Based AI | TYK2 inhibitor (NDI-034858) | $4B licensing deal with Takeda [35] | Phase III |
| BenevolentAI - Knowledge Graphs | Baricitinib for COVID-19 | AI-predicted drug repurposing [35] | Approved for COVID-19 |
| University of Chicago/Argonne - IDEAL | Ovarian cancer targets | Screening billions of molecules in hours [37] | Preclinical |
The performance data reveals consistent and substantial improvements across multiple dimensions of the drug discovery pipeline. AI-driven platforms demonstrate particular strength in the early discovery phases, with Exscientia and Insilico Medicine reporting the advancement of multiple compounds from target identification to clinical candidates in timeframes 60-70% shorter than traditional approaches [35]. Perhaps more significantly, early analyses suggest that AI-designed molecules progress to clinical trials at twice the rate of traditionally developed drugs and demonstrate substantially higher Phase 1 success rates of 80-90% compared to the industry standard of 40-50% [38]. This improvement in early-stage success rates represents a potential paradigm shift that could dramatically reduce the overall cost and time required to bring new oncology therapeutics to market.
The scalability of AI approaches is evidenced by projects like the University of Chicago and Argonne National Laboratory's IDEAL initiative, which leverages exascale supercomputing to screen billions of potential molecules in a matter of hours rather than months or years [37]. This computational advantage enables researchers to explore a much broader chemical and target space, including previously intractable target classes such as intrinsically disordered proteins that are highly relevant in oncology but have historically been considered undruggable [37].
Experimental Protocols and Methodologies
AI-Driven Target Identification and Validation
The AI-driven target identification process represents a fundamental departure from traditional hypothesis-driven approaches. Leading platforms employ diverse but complementary methodologies:
Knowledge Graph Integration (BenevolentAI): This approach integrates massive-scale biomedical data including scientific literature, patents, proteomics data, gene expression profiles, and clinical data using natural language processing and graph machine learning. The system identifies novel gene-disease-compound relationships by analyzing network topology and inferring hidden connections. For example, BenevolentAI's platform successfully predicted Baricitinib as a COVID-19 treatment by identifying its potential to inhibit viral endocytosis [35].
Multimodal Data Fusion (AstraZeneca's ABACO): This platform integrates imaging, histology, genomics, and clinical data from real-world evidence to identify predictive biomarkers and novel targets. The system employs transformer-based architectures similar to those used in natural language processing but adapted for multimodal biological data. In one implementation, the platform analyzed data from five Phase 3 prostate cancer trials and demonstrated a 9.2-14.6% relative improvement in predicting long-term outcomes compared to National Comprehensive Cancer Network risk stratification standards [38].
Phenotypic Screening (Recursion): This methodology systematically perturbs cell models using chemical or genetic interventions and employs computer vision and machine learning to detect subtle phenotypic changes indicative of therapeutic potential. The platform generates hundreds of terabytes of cellular imaging data which are processed using convolutional neural networks to quantify morphological features and predict mechanism of action [35].
The validation paradigm for AI-identified targets typically employs a multi-stage approach. For example, in a study identifying STK33 as a cancer target, researchers used an AI-driven screening strategy that integrated public databases and manually curated information. Target validation included in vitro studies demonstrating induction of apoptosis through STAT3 signaling pathway deactivation and cell cycle arrest at S phase, followed by in vivo validation showing decreased tumor size and induced necrotic areas [34].
Generative Molecular Design and Optimization
Generative AI approaches for molecular design have demonstrated remarkable efficiency improvements over traditional medicinal chemistry:
AI-Driven Molecular Design Workflow
Generative models including variational autoencoders (VAEs), generative adversarial networks (GANs), and more recently, transformer-based architectures explore chemical space more efficiently than traditional library-based approaches. For instance, Exscientia's "Centaur Chemist" platform combines AI algorithm creativity with human expert insight to automate the compound optimization process from target selection to clinical candidate [35]. The system employs a multi-parameter optimization strategy that simultaneously balances potency, selectivity, pharmacokinetic properties, and manufacturability constraints – a task that traditionally requires extensive iterative design cycles.
The integration of physical AI approaches with generative models represents a particularly promising advancement. Schrödinger's platform combines quantum mechanics, machine learning, and statistical modeling to achieve higher precision in molecular design [35]. This hybrid approach enables the prediction of binding affinities and conformational properties with accuracy sufficient to guide decision-making, as demonstrated by the successful development of TYK2 inhibitor NDI-034858, which was advanced to Phase III trials and secured a $4 billion licensing deal with Takeda [35].
High-Throughput Validation and Experimental Design
AI-driven approaches have revolutionized experimental validation through integrated design-make-test-analyze cycles:
Automated Laboratory Systems: Companies like Recursion have built automated robotic systems that can conduct millions of experiments weekly, generating standardized datasets for AI training. These systems integrate liquid handling, high-content imaging, and automated analysis to create closed-loop optimization systems [35].
Computational Superstructures: Projects like the IDEAL initiative leverage national supercomputing resources such as the Aurora exascale supercomputer at Argonne National Laboratory, which enables screening of billions of molecules in hours and simulation of thousands of complexes within days [37]. These resources are complemented by experimental facilities such as the Advanced Photon Source, which provides high-brightness X-rays for structural biology applications.
Multi-Agent Validation Systems: Innovative approaches like the ChemMAS system developed by Hangzhou Dianzi University create virtual teams of AI "experts" that specialize in different aspects of chemical optimization (catalysts, solvents, reagents) and engage in multi-round debates to reach consensus on optimal conditions. This system demonstrated 77.1% accuracy in catalyst prediction and 85.4% accuracy in solvent selection, significantly outperforming traditional approaches [39].
Signaling Pathways in AI-Driven Oncology Discovery
Multimodal AI in Oncology Discovery
The integration of multimodal artificial intelligence (MMAI) has enabled a more comprehensive understanding of oncogenic signaling pathways and their therapeutic targeting. MMAI approaches contextualize molecular features within anatomical and clinical frameworks, yielding more biologically plausible models of pathway dysfunction [38]. Several key examples illustrate this principle:
The TRIDENT machine learning model integrates radiomics, digital pathology, and genomics data from the Phase 3 POSEIDON study in metastatic non-small cell lung cancer (NSCLC). This multimodal approach identified a patient signature in >50% of the population that would obtain optimal benefit from a particular treatment strategy, with hazard ratio reductions ranging from 0.88-0.56 in non-squamous histology populations [38].
Pathomic Fusion, a multimodal fusion strategy combining histology and genomics in glioma and clear-cell renal-cell carcinoma datasets, outperformed the World Health Organization 2021 classification for risk stratification, demonstrating the clinical value of integrating multiple data modalities [38].
A pan-tumor analysis of 15,726 patients combined multimodal real-world data and explainable AI to identify 114 key markers across 38 solid tumors, which were subsequently validated in an external lung cancer cohort, demonstrating the generalizability of MMAI-derived biomarkers [38].
Research Reagent Solutions for AI-Driven Discovery
Table 3: Essential Research Reagents and Platforms for AI-Driven Oncology Discovery
| Research Tool Category | Specific Technologies/Platforms | Function in AI-Driven Discovery |
|---|---|---|
| AI Software Platforms | NVIDIA BioNeMo, Clara [40] | Training large biological foundation models for target and therapeutic discovery |
| Computational Infrastructure | Aurora Exascale Supercomputer [37], NVIDIA DGX SuperPOD [40] | Providing computational power for massive molecular screening and simulation |
| Data Integration Platforms | BenevolentAI Knowledge Graph [35], AstraZeneca ABACO [38] | Integrating multimodal data sources for target identification and validation |
| Generative Chemistry Tools | Exscientia Centaur Chemist [35], Insilico Medicine GENTRL [35] | De novo molecular design and multi-parameter optimization |
| Experimental Validation Systems | Recursion Phenotypic Screening Platform [35], Advanced Photon Source [37] | High-throughput experimental validation of AI-predictions |
| Multi-Agent Decision Systems | ChemMAS [39] | Multi-specialist AI system for reaction condition optimization |
| Clinical Trial Optimization | TRIDENT Machine Learning Model [38] | Patient stratification and biomarker identification for clinical trials |
The research infrastructure required for AI-driven oncology discovery represents a significant departure from traditional laboratory setups, with an increased emphasis on computational resources and data generation capabilities. The NVIDIA BioNeMo platform, for instance, enables researchers to train large foundation models on biological data, generating novel antibodies, nanobodies, and small molecules with unprecedented precision and speed [40]. Similarly, Lilly's AI factory, built on NVIDIA DGX systems, provides the computational infrastructure needed for genome-scale analysis and molecular simulation [40].
The integration of physical laboratory capabilities with computational approaches remains essential for validation. Facilities like the Advanced Photon Source at Argonne National Laboratory provide bright X-ray sources for determining atomic-level structures of targets and drug complexes, enabling the validation of AI-generated molecular designs [37]. Automated phenotypic screening platforms, such as those developed by Recursion, generate the high-quality, standardized datasets necessary to train predictive AI models on biological outcomes [35].
The comprehensive comparison between traditional and AI-driven approaches to oncology therapeutic discovery reveals a field undergoing rapid and transformative change. AI technologies are delivering substantial improvements across key metrics including discovery timeline compression, success rate improvement, and exploration of previously inaccessible target space. The 10⁵ times acceleration in molecular screening capabilities [36], coupled with the 2x improvement in Phase 1 success rates for AI-designed molecules [38], represents a fundamental shift in the economics and feasibility of oncology drug discovery.
Despite these promising advances, significant challenges remain. The clinical validation of AI-discovered targets and therapeutics still faces the same biological complexities and regulatory requirements as traditional approaches. Several high-profile setbacks, including the failure of BenevolentAI's Trk inhibitor BEN-2293 in Phase IIa trials and the termination of Recursion's REC-994 program for CCM disease, underscore that AI prediction does not guarantee clinical success [35]. The field must also address challenges related to data quality, model interpretability, and regulatory alignment as AI-driven approaches become more pervasive.
Looking forward, the convergence of AI with emerging experimental technologies promises to further accelerate progress. The integration of multimodal AI combining genomics, imaging, histopathology, and clinical data [38], the development of "physical AI" that incorporates biochemical principles into model architecture [40], and the creation of multi-agent systems that emulate scientific reasoning [39] represent the next frontier in oncology therapeutic discovery. As these technologies mature, they have the potential to not only accelerate existing processes but to fundamentally redefine the paradigm of drug discovery from serendipitous observation to predictable engineering, ultimately delivering more effective and personalized cancer treatments to patients in need.
Navigating the Hurdles: Data, Generalization, and Integration Challenges
Overcoming Data Scarcity and Ensuring Data Quality for Robust Models
The pursuit of high-performance catalysts is fundamental to advances in energy, pharmaceuticals, and green chemistry. Traditionally, catalyst development has been guided by empirical trial-and-error or theoretical simulations, approaches that are often slow, costly, and limited in their ability to navigate vast compositional and reaction spaces [2] [14]. The emergence of artificial intelligence (AI) and machine learning (ML) promises a paradigm shift, offering a data-driven path to accelerated discovery. However, the robustness of these AI models is critically dependent on the quality and quantity of the data used to train them. Data scarcity, often caused by the high cost and time-intensive nature of experimental catalysis research, and data quality issues, stemming from inconsistent experimental protocols or unstandardized data reporting, present significant bottlenecks [41] [42]. This guide objectively compares how traditional and AI-driven methodologies tackle these universal data challenges, providing researchers with a framework for building more reliable and predictive models in catalyst design.
Comparative Analysis: Traditional vs. AI-Driven Data Handling
The core difference between traditional and AI-driven approaches lies in how they acquire, manage, and leverage data. The following table summarizes their performance across key metrics relevant to data scarcity and quality.
Table 1: Performance Comparison of Traditional vs. AI-Driven Data Handling in Catalyst Development
| Performance Metric | Traditional Approach | AI-Driven Approach | Key Supporting Evidence |
|---|---|---|---|
| Data Acquisition Efficiency | Low; relies on sequential, manual experiments. | High; enabled by automated high-throughput synthesis & characterization [2]. | AI-EDISON & Fast-Cat platforms automate synthesis, generating larger, more robust datasets [2]. |
| Handling of Data Scarcity | Limited; struggles with small sample sizes. | Advanced; uses data augmentation & synthetic data generation [41]. | SMOTE generates synthetic minority-class samples, improving model performance on imbalanced data [41]. |
| Data Quality & Standardization | Often inconsistent; prone to manual entry errors and subjective interpretation. | Systematic; enforced through digital notebooks, automated ETL pipelines, and FAIR principles [42]. | Deloitte analysis shows FAIR data is critical for model reliability and regulatory confidence [42]. |
| Model Robustness & Predictive Accuracy | Variable; highly dependent on researcher expertise. | Higher and more consistent; excels in identifying complex, non-linear patterns from high-dimensional data [14]. | ML models can fit potential energy surfaces with exceptional accuracy and uncover mathematical laws for interpretability [2]. |
| Time-to-Solution | Long; often involves lengthy research cycles [2]. | Significantly reduced; accelerates screening and optimization loops [2] [18]. | AI-driven workflows can cut discovery time-to-market by 50% and reduce costs by 30% in R&D [18]. |
Experimental Protocols for Data-Centric Catalyst Development
To achieve the performance benchmarks outlined above, specific experimental methodologies are employed. The following protocols detail standardized workflows for generating high-quality data.
Protocol for AI-Driven High-Throughput Catalyst Screening
This protocol is designed to maximize data acquisition while ensuring consistency, directly addressing data scarcity and quality.
- Goal: Rapidly synthesize and screen a vast library of catalyst compositions to identify promising candidates for a target reaction (e.g., hydrogen evolution reaction).
- Materials & Setup:
- AI-EDISON or Fast-Cat Platform: An integrated system combining robotics for automated synthesis, a reactor for high-throughput testing, and online gas chromatography (GC) or mass spectrometry (MS) for performance evaluation [2].
- Precursor Libraries: Robotic dispensers and liquid handlers for precise delivery of metal salts and other precursors.
- Reaction Block: A multi-well reactor allowing parallel testing under controlled temperature and pressure.
- Procedure:
- Step 1 - Design of Experiment (DoE): The AI algorithm defines a set of catalyst compositions and synthesis conditions (e.g., temperature, precursor ratios) based on initial constraints and objectives.
- Step 2 - Automated Synthesis: Robotic arms execute the synthesis plan, preparing catalysts in the multi-well platform.
- Step 3 - High-Throughput Testing: Synthesized catalysts are evaluated in parallel in the reaction block. Performance data (e.g., conversion, selectivity) is automatically collected via online GC/MS.
- Step 4 - Data Integration and Model Retraining: All synthesis parameters and performance data are logged into a centralized database. The ML model is retrained on this new data to refine its predictions and propose the next set of optimal experiments.
- Step 5 - Iterative Loop: Steps 1-4 are repeated in a closed-loop fashion until a catalyst meeting the target performance criteria is identified [2].
Protocol for Mitigating Data Imbalance with SMOTE
This computational protocol addresses the common issue of imbalanced datasets, where high-performing catalysts are underrepresented.
- Goal: Balance a dataset of catalyst measurements to improve ML model accuracy in predicting high-performance candidates.
- Materials & Setup:
- Original Imbalanced Dataset: A collection of catalyst descriptors (e.g., composition, surface area) and a target property (e.g., Gibbs free energy of adsorption, ΔGH).
- Computational Environment: Python/R environment with libraries like
imbalanced-learn.
- Procedure:
- Step 1 - Data Preprocessing: Clean the data and split the catalysts into categories based on a performance threshold (e.g., 88 with |ΔGH| > 0.2 eV and 38 with |ΔGH| < 0.2 eV) [41].
- Step 2 - Identify Minority Class: Designate the class with fewer samples (e.g., high-performance catalysts with |ΔGH| < 0.2 eV) as the minority class.
- Step 3 - Apply SMOTE:
- For each sample in the minority class, find its k-nearest neighbors.
- Synthesize new examples by randomly interpolating between the original sample and its neighbors.
- Step 4 - Dataset Validation: The result is a balanced dataset with even distribution between classes, which is then used to train ML models like XGBoost or Random Forest, leading to improved predictive performance for the previously underrepresented class [41].
Visualizing Workflows and Logical Relationships
AI-Driven Closed-Loop Catalyst Development Workflow
This diagram illustrates the integrated, data-centric workflow that enables AI-driven platforms to efficiently overcome data scarcity.
Data Quality Management Process for AI-Ready Data
This diagram outlines the systematic process required to ensure data quality, a foundational element for robust AI models.
The Scientist's Toolkit: Key Research Reagent Solutions
Building robust AI models requires both computational and physical tools. The following table details essential solutions for a modern catalysis research lab.
Table 2: Essential Research Reagent Solutions for AI-Driven Catalyst Development
| Research Solution | Function | Application in Catalysis |
|---|---|---|
| Automated High-Throughput Synthesis Platform | Robotic system for parallelized, hands-off preparation of catalyst libraries. | Enables rapid generation of large, consistent experimental datasets to overcome data scarcity [2]. |
| SMOTE & Advanced Oversampling Algorithms | Computational techniques to generate synthetic data for underrepresented classes. | Balances imbalanced catalyst datasets, improving model accuracy for predicting high-performance materials [41]. |
| FAIR Data Management Platform | Implements Findable, Accessible, Interoperable, and Reusable data principles. | Ensures data quality, reproducibility, and seamless integration from multiple sources for reliable AI models [42]. |
| AI-Driven Data Cleansing Tools | Software that uses AI to detect anomalies, correct errors, and standardize data formats. | Automates the data preparation phase, reducing manual effort and improving the integrity of training data [42]. |
| Multi-Modal Characterization Data Integrator | System that correlates data from different techniques (e.g., microscopy, spectroscopy). | Provides comprehensive structural-property relationship insights, enriching dataset features for ML models [2]. |
Artificial intelligence is reshaping scientific discovery, particularly in high-stakes fields like catalyst development and drug discovery. However, the most advanced AI systems remain 'black boxes'—their internal decision-making processes are opaque, even to their creators [43] [44]. This opacity presents a fundamental challenge for research applications where understanding the 'why' behind a prediction is as crucial as the prediction itself.
The black box problem stems from the inherent complexity of modern AI architectures, especially deep learning models with multiple hidden layers containing millions of parameters [43] [45]. As these models make increasingly consequential decisions—from diagnosing diseases to designing novel catalysts—the lack of transparency complicates validation, trust, and accountability [46] [47]. In pharmaceutical research, where AI spending is expected to reach $3 billion by 2025, this interpretability gap becomes a critical barrier to adoption [48].
This guide examines strategies for AI model interpretability, comparing traditional and AI-driven approaches within catalyst development. We evaluate interpretability techniques through both technical capabilities and practical research applications, providing scientists with a framework for selecting appropriate methods for their specific research contexts.
The Black Box Challenge in Scientific AI
Defining the Interpretability Problem
The AI black box problem refers to the lack of transparency in how machine learning models, particularly deep learning systems, arrive at their conclusions [43] [47]. Unlike traditional software with predefined rules, these models learn complex patterns from data through processes that are difficult to trace or interpret. The challenge is most pronounced in deep neural networks, where data transformations across hundreds or thousands of layers create representations that don't readily map to human-understandable concepts [45].
In scientific contexts, this opacity manifests differently across research paradigms. Traditional catalyst development relies on established principles like linear free energy relationships (Hammett equation, Brønsted catalysis law) that provide transparent, interpretable relationships between molecular structure and activity [1]. In contrast, AI-driven approaches can identify complex, non-linear patterns across high-dimensional parameter spaces but often lack explanatory mechanisms, creating a tension between predictive power and interpretability [1].
Consequences for Research and Development
The interpretability gap has tangible implications for scientific progress:
- Validation Challenges: In healthcare, 94% of 516 machine learning studies failed to pass even the first stage of clinical validation tests, raising questions about AI reliability without interpretability [47].
- Bias Amplification: AI models can perpetuate and amplify biases present in training data. Amazon's recruiting engine famously penalized resumes containing the word "women's," reflecting biases in historical hiring data [43].
- Accountability Gaps: When AI systems make errors—such as misdiagnosing conditions or recommending suboptimal catalysts—the opaque decision process complicates responsibility assignment and error correction [45].
Comparative Analysis of Interpretability Strategies
Taxonomy of Interpretability Approaches
Researchers have developed numerous techniques to address the black box problem, each with distinct mechanisms and applications. The table below compares prominent interpretability methods:
Table 1: Comparison of AI Model Interpretability Approaches
| Method | Category | Mechanism | Research Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| LIME (Local Interpretable Model-agnostic Explanations) | Model-Agnostic | Approximates black box models with local interpretable models | Catalyst performance prediction, reaction optimization | Works with any model; provides local explanations | Explanations can be unstable; computationally expensive [45] [47] |
| SHAP (SHapley Additive exPlanations) | Model-Agnostic | Game theory-based feature importance scores | Molecular property prediction, materials design | Theoretical foundation; consistent explanations | Computationally intensive; complex for non-experts [45] [47] |
| Attention Mechanisms | Model-Specific | Highlights input regions influencing decisions | Chemical literature analysis, reaction prediction | Intuitive visualization; built into model architecture | Correlation ≠ causation; may highlight features without causal relationships [45] [47] |
| Counterfactual Explanations | Model-Agnostic | Shows how input changes would alter outputs | Catalyst design, molecular optimization | Actionable insights; intuitive presentation | Doesn't reveal original decision process; multiple possible counterfactuals [47] |
| Inherently Interpretable Models | Model Design | Uses transparent architectures by design | Preliminary screening, regulatory applications | Built-in transparency; no additional explanation needed | Often sacrificed predictive power for interpretability [47] |
Experimental Validation in Catalyst Design
The Catal-GPT framework provides a case study in implementing interpretability methods for catalyst development. This AI assistant interacts with researchers to optimize catalyst formulations for reactions like oxidative coupling of methane (OCM) [9]. The experimental protocol below illustrates how interpretability techniques can be integrated into AI-driven research:
Table 2: Experimental Protocol for Evaluating Interpretability in Catalyst AI
| Stage | Methodology | Interpretability Assessment |
|---|---|---|
| Data Preprocessing | Collection of catalyst synthesis, characterization, and application data from literature; cleaning and encoding for model input | Data provenance documentation; bias assessment in training data [9] |
| Model Training | Fine-tuning of Qwen2-7B LLM on specialized catalyst database; transfer learning from general chemical knowledge | Attention visualization to identify which training examples most influence specific predictions [9] |
| Knowledge Extraction | Model questioned on specific catalyst parameters (e.g., calcination temperature for La2O3/CaO systems) | Accuracy quantification (92% in tested cases); top-k recall analysis by question type [9] |
| Prediction Validation | Experimental testing of AI-proposed catalyst formulations compared to traditional design approaches | SHAP analysis to identify features driving predictions; counterfactual testing of proposed catalysts [9] |
The experimental results revealed significant variation in interpretability across question types. While the model achieved 80% recall (top-1) and 100% recall (top-7) for theoretical calculation methods, it showed lower performance (plateauing at 80% by top-7) for catalyst preparation details, highlighting domain-specific interpretability challenges [9].
Interdisciplinary Framework for Interpretable AI
Workflow Integration Strategy
Successfully addressing the black box problem requires integrating interpretability throughout the research workflow. The diagram below illustrates this integrated approach:
Integrated Interpretability Workflow for Scientific AI
This framework emphasizes that interpretability isn't a standalone phase but integrated throughout the AI research pipeline. Different interpretability methods serve complementary roles: model-agnostic approaches like SHAP provide post-hoc explanations for complex models, while inherently interpretable architectures offer built-in transparency where predictive requirements allow [45] [47].
Implementing effective interpretability strategies requires both technical tools and methodological awareness. The following table catalogues key resources mentioned in experimental studies:
Table 3: Research Reagent Solutions for AI Interpretability
| Tool/Category | Specific Examples | Function in Interpretability | Research Applications |
|---|---|---|---|
| Model-Agnostic Libraries | SHAP, LIME, ELI5 | Post-hoc explanation generation for any model | Feature importance analysis in catalyst design [45] [47] |
| Visualization Tools | Saliency maps, Attention heatmaps, Feature importance graphs | Visual representation of model focus areas | Identifying critical molecular descriptors in QSAR studies [45] |
| Specialized AI Platforms | IBM AI Explainability 360, Google Model Interpretability, Anthropic's Interpretability Research | Pre-built algorithms and frameworks for explainability | Regulatory compliance; model debugging in pharmaceutical applications [46] [49] |
| Benchmarking Datasets | Catalyst-specific corpora, Molecular property benchmarks | Standardized evaluation of interpretability methods | Comparing explanation accuracy across different catalyst classes [9] [1] |
| Traditional Modeling Approaches | Linear regression, Decision trees, Rule-based systems | Baseline interpretable models for comparison | Establishing performance-interpretability tradeoff benchmarks [45] [47] |
Future Directions and Research Opportunities
Emerging Approaches in Interpretable AI
The field of AI interpretability is evolving rapidly, with several promising research directions:
- Causal Inference Integration: Moving beyond correlation to understand causal relationships in AI decision-making [47].
- Dynamic Prompt Engineering: Frameworks that deeply couple the semantic parsing capabilities of large language models with physical models like density functional theory calculations [9].
- Standardized Knowledge Graphs: Enhancing accuracy of unstructured experimental data extraction through self-supervised contrastive learning mechanisms [9].
- Human-Centric Explanation Design: Developing explanations tailored to different stakeholder needs, from laboratory researchers to regulatory officials [47].
Strategic Implementation Recommendations
For research organizations navigating the transition toward AI-driven approaches, we recommend:
- Match Interpretability Methods to Research Context: High-stakes applications like clinical trial optimization warrant more rigorous interpretability approaches than preliminary screening [50].
- Prioritize Human-AI Collaboration: Systems like Catal-GPT demonstrate the power of conversational interfaces that allow researchers to interrogate AI suggestions directly [9].
- Embrace Hybrid Modeling: Combining AI with traditional theoretical frameworks (e.g., DFT calculations) creates more trustworthy and interpretable systems [9] [1].
- Invest in Interpretability Infrastructure: As the XAI market grows (projected to reach $20.74 billion by 2029), institutional investment in interpretability tools and expertise will yield increasing returns [46].
The black box problem represents a fundamental challenge in AI-driven scientific discovery, particularly in fields like catalyst development and pharmaceutical research. While techniques like LIME, SHAP, and attention mechanisms provide partial solutions, no single approach fully resolves the tension between model complexity and interpretability. The most promising path forward involves context-aware integration of multiple interpretability strategies throughout the research workflow, combined with methodological transparency about the limitations of each approach.
As AI capabilities advance—with systems projected to reach human-level performance in certain scientific domains within the coming decade—addressing the interpretability gap becomes increasingly urgent [44]. By adopting the comparative framework presented in this guide, researchers can make informed decisions about implementing interpretable AI systems that balance predictive power with explanatory capability, ultimately accelerating scientific discovery while maintaining rigorous standards of validation and trust.
Catalyst deactivation remains a fundamental challenge in industrial catalysis, compromising performance, efficiency, and sustainability across numerous chemical processes. Traditional approaches to mitigating deactivation have relied heavily on empirical observations and trial-and-error experimentation, often resulting in lengthy development cycles and suboptimal solutions. The principal deactivation pathways—including coking, poisoning, thermal degradation, and mechanical damage—consume substantial manpower and material resources while introducing uncertainty into research outcomes [2] [28]. In industrial settings, catalyst deactivation can occur rapidly, as in fluidized catalytic cracking (FCC), or gradually over several years, as in NH₃ synthesis, but its economic impact is consistently significant [28].
The emergence of artificial intelligence (AI) and machine learning (ML) is sharply transforming this research paradigm, offering powerful new tools to tackle the highly complex issues within every aspect of catalyst design and operation. AI provides unique advantages in tackling the high-dimensionality of the search space consisting of catalyst composition, structure, reactants, and synthesis conditions [2]. This article provides a comprehensive comparison between traditional and AI-driven approaches for mitigating catalyst deactivation, offering researchers a structured framework for evaluating these methodologies across key performance parameters.
Traditional Approaches to Understanding and Mitigating Catalyst Deactivation
Established Deactivation Mechanisms and Countermeasures
Traditional catalyst development has established a solid understanding of primary deactivation pathways and corresponding mitigation strategies:
Coking/Fouling: Carbonaceous deposits block active sites and pores, typically addressed through periodic oxidative regeneration (burning with air/O₂) [28]. This remains the most prevalent deactivation mechanism in industrial processes involving organic compounds.
Poisoning: Strong chemisorption of impurities (e.g., sulfur, heavy metals) necessitates feedstock purification or the development of poison-resistant catalyst formulations [28].
Thermal Degradation/Sintering: High temperatures cause crystallite growth and surface area reduction, mitigated through improved thermal stability in catalyst design [28].
Mechanical Damage: Attrition and crushing require enhanced structural integrity through binders and improved reactor design [28].
Conventional Experimental Methodologies
Traditional experimental protocols for studying deactivation involve standardized accelerated aging tests:
Long-term Stability Testing: Catalysts are evaluated under realistic process conditions for extended durations (often thousands of hours) with periodic activity measurements.
Accelerated Deactivation Protocols: Exposure to extreme conditions (higher temperature, concentrated poisons) to simulate extended operation in shortened timeframes.
Post-mortem Characterization: Techniques including temperature-programmed oxidation (TPO) for coke analysis, chemisorption for active site quantification, and electron microscopy for structural assessment.
Regeneration Procedure Optimization: Systematic evaluation of burn-off parameters (temperature, O₂ concentration, space velocity) to restore activity while minimizing thermal damage.
The limitations of these traditional approaches include their resource-intensive nature, limited exploration of parameter space, and slow knowledge accumulation cycles, often requiring years to develop stable catalyst formulations for new processes [2] [3].
AI-Driven Frameworks for Predictive Stability and Deactivation Mitigation
Fundamental Shift in Research Paradigm
AI and machine learning introduce a transformative approach to addressing catalyst deactivation by leveraging data-driven pattern recognition and predictive modeling. The core advantage lies in ML algorithms' ability to process massive computational and experimental datasets to identify complex, non-linear relationships between catalyst properties, operating conditions, and deactivation behavior [2] [13].
Three key ML paradigms are particularly relevant for deactivation studies:
- Supervised Learning: Trains models on labeled datasets (e.g., catalysts with known deactivation rates) to predict longevity from molecular descriptors [13] [14].
- Unsupervised Learning: Identifies hidden patterns and groupings in unlabeled deactivation data, enabling discovery of previously unrecognized deactivation mechanisms [13].
- Reinforcement Learning: Optimizes catalyst regeneration protocols through iterative virtual testing, minimizing experimental requirements [3].
Machine Learning Algorithms for Stability Prediction
Different ML algorithms offer distinct advantages for various aspects of deactivation prediction:
Random Forest ensembles multiple decision trees to handle high-dimensional descriptor spaces and provide feature importance rankings for deactivation factors [13] [14].
Neural Networks capture complex non-linear relationships between catalyst composition, structure, and deactivation behavior, particularly effective with large, diverse datasets [13].
Gaussian Process Regression provides uncertainty quantification alongside predictions, valuable for assessing reliability of longevity forecasts [51].
Symbolic Regression (e.g., via SISSO algorithm) discovers mathematically interpretable expressions connecting catalyst features to deactivation rates, offering physical insights alongside predictions [14].
Comparative Analysis: Traditional vs. AI-Driven Approaches
Table 1: Comparison of Traditional and AI-Driven Approaches to Catalyst Deactivation Mitigation
| Aspect | Traditional Approaches | AI-Driven Approaches |
|---|---|---|
| Time Requirements | Years for development cycles [2] | Months to years [2] |
| Experimental Throughput | Limited by manual operations [2] | High-throughput automated systems [2] |
| Parameter Optimization | One-factor-at-a-time testing [3] | Multi-dimensional simultaneous optimization [3] |
| Deactivation Prediction | Based on known mechanisms & analogs [28] | Data-driven from complex feature interactions [14] |
| Mechanistic Insight | Direct but limited to characterized systems [28] | Pattern-based but sometimes black-box [13] |
| Resource Requirements | High laboratory resource consumption [2] | High computational resources [3] |
| Regeneration Protocol Development | Empirical optimization [28] | Model-guided optimization [28] |
Table 2: Performance Comparison for Specific Catalyst Systems
| Catalyst System | Deactivation Mechanism | Traditional Longevity | AI-Optimized Longevity | Key Improvement Factors |
|---|---|---|---|---|
| Zeolite Catalysts | Coking, dealumination [28] | 12-24 months [28] | 30-40 months [28] | Composition optimization, regeneration parameters |
| Single-Atom Catalysts | Sintering, poisoning [52] | Weeks to months [52] | 3-6x improvement [52] | Support interaction optimization |
| Pd-based Catalysts | SO₂ poisoning, coking [28] | 60% activity loss in 100h [28] | 25% activity loss in 100h [28] | Poison-resistant formulations |
Experimental Protocols for AI-Enhanced Deactivation Studies
Data Generation and Curation Framework
High-quality, standardized data forms the foundation of effective AI models for deactivation prediction:
Accelerated Aging Data Collection:
- Conduct parallel deactivation experiments across multiple catalysts under controlled conditions
- Measure activity, selectivity, and characterization parameters at regular intervals
- Employ standardized protocols to ensure data consistency and comparability [14]
Multi-scale Descriptor Calculation:
- Compute atomic-level descriptors (electronic structure, coordination numbers)
- Extract morphological features (surface area, porosity, particle size distribution)
- Process operational parameters (temperature, pressure, feed composition) [51]
Data Management and Standardization:
- Implement FAIR (Findable, Accessible, Interoperable, Reusable) data principles
- Apply consistent metadata schemas for experimental conditions
- Utilize automated data capture from analytical instruments [51]
Machine Learning Model Development Workflow
AI Model Development Workflow
Integrated AI-Driven Platforms for Autonomous Catalyst Development
The most advanced applications of AI for deactivation mitigation involve closed-loop autonomous systems that integrate prediction, synthesis, and testing:
Closed-Loop AI Catalyst Development
Systems such as AI-EDISON and Fast-Cat exemplify this approach, combining ML algorithms with high-throughput synthesis and characterization technologies to form autonomous discovery platforms [2]. These systems can conduct the necessary long-term experiments to study slow deterioration processes while maximizing information content through intelligent experimental design [53].
The FHI Berlin self-driving laboratory represents cutting-edge implementation, specifically targeting catalyst deactivation studies through AI-guided long-term experimentation [53]. Such systems progressively improve their understanding of deactivation mechanisms with each experimental cycle, accelerating the development of more stable catalyst formulations.
The Researcher's Toolkit: Essential Solutions for AI-Enhanced Deactivation Studies
Table 3: Essential Research Tools for AI-Driven Catalyst Deactivation Studies
| Tool Category | Specific Solutions | Function in Deactivation Research |
|---|---|---|
| AI/ML Platforms | Random Forest, Neural Networks, Gaussian Process Regression [13] [14] | Predict deactivation rates, identify key descriptors, optimize regeneration |
| Automation Systems | High-throughput robotic synthesis [2], Automated testing reactors [2] | Generate consistent deactivation data, enable long-term unsupervised studies |
| Characterization | In-situ spectroscopy, Automated microscopy [2] | Provide real-time deactivation monitoring, structural changes during operation |
| Data Management | Laboratory Information Management Systems (LIMS) [3], FAIR data platforms [51] | Ensure data quality, standardization, and interoperability for ML models |
| Computational Tools | Density Functional Theory (DFT) [13], Microkinetic modeling [51] | Generate training data, provide physical insights for interpretable AI |
| Analysis Frameworks | SHAP (SHapley Additive exPlanations) [14], Symbolic regression [14] | Interpret ML models, extract mathematical relationships for deactivation |
Future Perspectives and Implementation Guidelines
The integration of AI into catalyst deactivation research continues to evolve, with several emerging trends shaping its future trajectory:
AI Agents for Decision-Making: Development of autonomous systems capable of planning and executing multi-step deactivation studies with minimal human intervention [19].
Advanced Feature Extraction: Improved techniques for processing complex characterization data (spectroscopy, microscopy) to identify subtle structural changes preceding deactivation [2].
Multi-modal Data Integration: Combining operational data with characterization results and computational simulations to build comprehensive digital twins of catalyst aging [2].
Explainable AI (XAI): Enhanced model interpretability through techniques like SHAP analysis and symbolic regression, bridging the gap between data-driven predictions and physical understanding [14].
For research teams implementing AI approaches for deactivation studies, a phased strategy is recommended:
Begin with supervised learning on well-characterized catalyst systems to establish baseline predictive capabilities
Implement automated data capture and management to ensure data quality and accessibility
Progress toward closed-loop systems that integrate prediction, synthesis, and testing for autonomous optimization
Focus on interpretability to ensure AI insights translate to fundamental understanding and not just empirical correlations
The transformation from traditional to AI-enhanced approaches for mitigating catalyst deactivation represents a paradigm shift in catalysis research. While traditional methods provide fundamental mechanistic understanding and remain essential for validation, AI-driven approaches offer unprecedented capabilities for navigating complex parameter spaces and accelerating the development of stable catalyst systems. The most effective strategies leverage the strengths of both approaches, combining data-driven pattern recognition with physical insights to create a comprehensive understanding of deactivation phenomena.
As AI technologies continue to mature—with advances in autonomous laboratories, explainable AI, and multi-modal data integration—their impact on catalyst longevity and stability is expected to grow substantially. Research organizations that strategically integrate these approaches position themselves to develop more durable, efficient, and sustainable catalytic processes, ultimately translating to improved economic and environmental outcomes across the chemical industry.
The fields of catalyst design and drug discovery are undergoing a profound transformation, shifting from traditional, intuition-driven methodologies to intelligence-guided, data-driven processes. This paradigm shift, powered by Artificial Intelligence (AI), is addressing long-standing challenges in retrosynthetic design, catalyst design, reaction development, and autonomous experimentation [1]. Historically, progress in these areas relied heavily on fundamental principles, experimental ingenuity, and serendipity, with classical models providing elegant but simplified structure-activity relationships based on limited datasets. The integration of AI is now enabling researchers to explore high-dimensional chemical spaces, optimize reaction conditions, and accelerate novel reaction discovery with unparalleled efficiency and precision [1]. This article provides a comprehensive comparison between traditional and AI-driven development approaches, underpinned by experimental data and detailed protocols, to illuminate the transformative potential of this integration for researchers, scientists, and drug development professionals.
Comparative Analysis: Traditional vs. AI-Driven Development
The following tables quantify the performance differences between traditional and AI-driven approaches across key metrics in catalyst and drug discovery.
Table 1: Comparative Performance in Catalyst Discovery and Optimization
| Performance Metric | Traditional Approach | AI-Driven Approach | Experimental Validation |
|---|---|---|---|
| Exploration Efficiency | Limited by human capacity; sequential testing | High-throughput screening of 900+ chemistries and 3,500+ tests in 3 months [4] | CRESt platform discovery of a multielement catalyst with 9.3-fold improvement in power density per dollar over pure Pd [4] |
| Catalyst Performance | Incremental improvements based on known elements | Discovery of novel multi-element compositions [4] | Record power density in a direct formate fuel cell with one-fourth the precious metals [4] |
| Parameter Space | Constrained by researcher experience and predefined hypotheses | Vast, non-linear space exploration with dynamic adjustment [20] | AI-HTE integration addresses challenges of vast parameter spaces and non-linear relationships [20] |
Table 2: Comparative Performance in Drug Discovery
| Performance Metric | Traditional Approach | AI-Driven Approach | Experimental Validation |
|---|---|---|---|
| Discovery Timeline | ~5 years to candidate [31] | As little as 18 months to Phase I trials [31] | Insilico Medicine's idiopathic pulmonary fibrosis drug [31] |
| Compound Efficiency | Thousands of synthesized compounds per program [31] | 10x fewer compounds synthesized; 70% faster design cycles [31] | Exscientia's CDK7 inhibitor candidate from 136 synthesized compounds [31] |
| Development Cost | ~$2.6 billion per new drug [48] | Up to 40% cost reduction in discovery [48] | AI-enabled workflows reduce time and cost to preclinical candidate stage [48] |
Experimental Protocols: Validating AI Predictions
Protocol 1: AI-Guided High-Throughput Catalyst Discovery
This protocol is adapted from the CRESt (Copilot for Real-world Experimental Scientists) platform developed by MIT researchers [4].
- Objective: To discover and optimize a multielement fuel cell catalyst with high power density and reduced precious metal content.
- AI Prediction Phase:
- Step 1: The AI model is given a objective (e.g., "maximize power density per cost for a formate fuel cell catalyst") and access to a knowledge base of scientific literature and existing experimental data.
- Step 2: Using natural language processing, the system searches scientific papers for descriptions of elements or precursor molecules with potentially useful properties.
- Step 3: An active learning model, incorporating both literature knowledge and Bayesian optimization, suggests an initial set of promising material recipes from a vast chemical space.
- Experimental Validation Phase:
- Step 4 (High-Throughput Synthesis): A liquid-handling robot prepares catalyst precursors based on the AI's recipes. A carbothermal shock system then rapidly synthesizes the materials.
- Step 5 (Automated Characterization): The synthesized materials are automatically characterized using techniques such as electron microscopy and X-ray diffraction.
- Step 6 (Performance Testing): An automated electrochemical workstation tests the catalytic activity, selectivity, and stability of each material.
- Step 7 (Iterative Learning): Results from characterization and testing are fed back to the AI model. The model uses this multimodal feedback (experimental data, literature, and human input) to refine its hypotheses and design the next round of experiments. This loop continues until a performance target is met [4].
Protocol 2: AI-Driven De Novo Drug Design
This protocol is based on the workflows of leading AI-driven drug discovery platforms like Exscientia and Insilico Medicine [31].
- Objective: To design a novel small-molecule drug candidate targeting a specific disease, optimizing for efficacy and pharmacokinetics.
- AI Prediction Phase:
- Step 1 (Target Identification): AI algorithms analyze vast amounts of biological data (genomics, proteomics) to identify novel, druggable disease targets.
- Step 2 (Generative Molecular Design): Generative AI models, trained on vast chemical libraries and structure-activity relationship data, propose novel molecular structures that are predicted to bind to the target and have favorable properties (potency, selectivity, ADME).
- Step 3 (In Silico Screening): The generated molecules are virtually screened against the target using deep learning and physics-based simulations to predict binding affinity and reduce undesirable off-target interactions.
- Experimental Validation Phase:
- Step 4 (Synthesis): The top-ranking AI-designed molecules are synthesized. Platforms with integrated "AutomationStudio" labs use robotics to accelerate this step.
- Step 5 (In Vitro Testing): Compounds are tested in biochemical and cell-based assays to validate target engagement and efficacy.
- Step 6 (Ex Vivo/Patient-First Validation): For oncology, compounds may be tested on patient-derived tissue samples (e.g., Allcyte platform acquired by Exscientia) to assess translational relevance in a human disease context [31].
- Step 7 (Lead Optimization): Data from experimental testing is used to retrain the AI models, which then design improved molecules in the next iterative cycle, compressing the traditional design-make-test-analyze loop [31].
Workflow Visualization: AI-Integrated Discovery
The following diagram illustrates the continuous, iterative cycle of an AI-integrated discovery platform, as implemented in systems like CRESt and AI-driven drug discovery platforms.
AI-Integrated Discovery Workflow
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Reagent Solutions and Platforms for AI-Integrated Research
| Item / Platform | Function in AI-Driven Workflow |
|---|---|
| CRESt (MIT) | A comprehensive platform that uses multimodal AI (literature, experimental data) and robotics for high-throughput materials discovery and optimization [4]. |
| Centaur Chemist (Exscientia) | An AI-driven drug design platform that integrates algorithmic molecule generation with human expertise for iterative compound optimization [31]. |
| High-Throughput Electrochemical Workstation | Enables automated, parallel testing of catalyst activity, selectivity, and stability, generating the large, consistent datasets required for AI training [4] [20]. |
| Liquid-Handling Robots | Automates the precise preparation and synthesis of catalyst precursors or drug compound libraries based on AI-generated recipes, ensuring reproducibility and speed [4] [31]. |
| SISSO Algorithm | An explainable machine learning algorithm used to identify key physical descriptors that govern material performance, bridging the gap between AI prediction and scientific understanding [20]. |
| Patient-Derived Biological Samples | Provides ex vivo disease models (e.g., patient tumor samples) for validating AI-designed drug candidates, enhancing translational relevance and clinical success probability [31]. |
The integration of AI predictions with rigorous experimental validation represents a fundamental shift in scientific research methodology. The quantitative data and experimental protocols presented herein demonstrate that AI-driven approaches consistently outperform traditional methods in terms of speed, efficiency, and the ability to discover novel, high-performing catalysts and drug candidates. While human researchers remain indispensable for defining objectives, providing domain expertise, and interpreting complex results [4], the synergy between AI's exploration power and high-throughput experimental validation is undeniable. This hybrid "centaur" model, leveraging the strengths of both human and machine intelligence, is poised to accelerate the discovery of solutions to some of the most challenging problems in energy and medicine.
By the Numbers: Quantifying the Impact of AI on Catalyst R&D
The field of catalyst development is undergoing a profound transformation, moving from traditional trial-and-error experimentation to data-driven design powered by artificial intelligence. This shift is particularly critical in demanding sectors such as pharmaceuticals and energy, where the acceleration of discovery timelines and the reduction of development costs are paramount. Traditional methods, while foundational, often involve extensive, sequential laboratory work that is both time-consuming and resource-intensive. In contrast, AI-driven approaches leverage machine learning, robotic automation, and multimodal data integration to explore chemical spaces more efficiently and intelligently. This guide provides a objective, data-backed comparison of these two paradigms, focusing on quantitative metrics of development timelines and costs. It is designed to equip researchers, scientists, and development professionals with a clear understanding of the operational and financial implications of adopting AI-driven workflows, framed within the broader thesis of comparing traditional and modern catalyst development approaches.
Methodology of Comparison
To ensure a fair and accurate comparison, the methodologies of both traditional and AI-driven catalyst development were analyzed based on a review of current literature and published experimental protocols. The comparison focuses on a hypothetical, yet representative, catalyst discovery project aimed at identifying a novel multi-element catalyst for an application such as a fuel cell.
Traditional Catalyst Development Workflow
The traditional approach is largely linear and human-centric. It relies on a researcher's intuition and deep domain knowledge to formulate hypotheses based on a review of existing scientific literature. Experiments are then designed manually, one at a time or in small batches. The process involves the sequential steps of manual precursor preparation and synthesis, followed by characterization (e.g., X-ray diffraction, electron microscopy) and performance testing (e.g., electrochemical analysis). The data from each experiment is analyzed, and the results inform the next, singular hypothesis and experiment. This cycle repeats until a material meeting the target specifications is identified. A significant challenge within this workflow is maintaining reproducibility, as subtle variations in manual processing can introduce inconsistencies that are difficult to debug, often requiring painstaking, manual investigation [4].
AI-Driven Catalyst Development Workflow
The AI-driven approach, exemplified by platforms like MIT's CRESt (Copilot for Real-world Experimental Scientists), is an iterative, closed-loop system. It begins with the AI model ingesting a wide range of information, including scientific literature, existing databases, and chemical knowledge, to create a knowledge-embedded search space. This space is then refined using techniques like principal component analysis. An active learning loop, often guided by Bayesian optimization, selects the most promising recipe or experiment to perform next based on all accumulated data. A key differentiator is the use of robotic equipment for high-throughput synthesis and testing, executing the AI's suggested experiments autonomously. The resulting multimodal data—from performance tests, automated characterization, and even computer vision monitoring of the experiments themselves—is fed back into the AI models. This data not only refines the search for the next experiment but also, through integration with large language models, augments the system's knowledge base. This creates a continuous cycle of proposal, testing, and learning that dramatically accelerates the discovery process [4].
Comparative Experimental Protocol
For a head-to-head comparison, the following unified experimental protocol is proposed, applicable to both methodologies:
- Objective: Discover a multi-element (3+ elements) catalyst that exceeds a target performance metric (e.g., power density in a direct formate fuel cell).
- Defined Search Space: A set of permissible precursor elements and a range of processing parameters (e.g., temperature, pressure) will be established for both approaches.
- Performance Benchmark: The primary success metric will be the improvement in power density per dollar of catalyst cost compared to a baseline pure palladium catalyst.
- Termination Condition: The experiment will be concluded when a catalyst meeting or exceeding the target performance metric is identified and validated.
The key variable is the process each method uses to navigate from the starting search space to the final successful catalyst.
Comparative Data Analysis
The quantitative differences between traditional and AI-driven development are stark, impacting both the pace of discovery and the associated costs. The data reveals that AI-driven methods are not merely incremental improvements but represent a fundamental shift in efficiency.
Development Timeline and Throughput Comparison
The following table summarizes the key differences in the speed and scale of experimental work. AI-driven workflows leverage automation to achieve a level of throughput that is impractical for traditional manual methods.
Table 1: Timeline and Throughput Metrics
| Metric | Traditional Approach | AI-Driven Approach | Data Source / Context |
|---|---|---|---|
| Experimental Cycle Time | Weeks to months per iterative cycle | Days to hours per iterative cycle | [4] |
| Number of Chemistries Explored | Limited by manual effort (e.g., 10s-100s) | High-throughput (e.g., 900+) | [4] (900+ chemistries explored by AI) |
| Number of Tests Performed | Limited by manual effort | Large-scale automated testing (e.g., 3,500+) | [4] (3,500+ tests performed by AI) |
| Project Duration to Discovery | Can span several years | Can be compressed to months | [4] (Discovery achieved in ~3 months) |
Development Cost and Resource Analysis
The cost structures of these two approaches are fundamentally different. Traditional methods incur high and variable labor costs, while AI-driven methods involve significant upfront investment in technology and infrastructure but can lead to substantial long-term savings and higher value outcomes.
Table 2: Cost and Resource Metrics
| Metric | Traditional Approach | AI-Driven Approach | Data Source / Context |
|---|---|---|---|
| Primary Cost Driver | Highly-skilled researcher labor & materials | AI infrastructure, compute power, & data management | [54] [55] |
| Cost Predictability | Linear and more predictable per experiment | Non-linear; initial setup high, then lower marginal cost per experiment | [54] |
| Reported Efficiency Gain | Baseline | 30-50% productivity improvements reported in applied AI settings | [56] [57] |
| Return on Investment (ROI) | Not specifically quantified for research | Top performers see ~$10.30 return per dollar invested in GenAI | [57] |
| Value Demonstrated | Incremental improvements | Record-breaking performance (e.g., 9.3x improvement in power density per $) | [4] |
The Scientist's Toolkit: Research Reagent Solutions
The transition to AI-driven science relies on a new class of "reagents"—the software, hardware, and data solutions that enable autonomous discovery. The following table details the key components of a modern, AI-ready materials science toolkit.
Table 3: Essential Components for an AI-Driven Research Laboratory
| Item | Function in Research |
|---|---|
| Multimodal AI Platform (e.g., CRESt) | Integrates diverse data sources (literature, experimental data, images) to plan and optimize experiments using natural language commands [4]. |
| Active Learning/Bayesian Optimization Software | The core algorithm that selects the most informative experiment to perform next, maximizing the learning per trial [4]. |
| Liquid-Handling Robot | Automates the precise preparation of material precursors, enabling high-throughput and reproducible synthesis [4]. |
| Automated Characterization Suite | Includes instruments like electron microscopes and X-ray diffractometers configured for automated analysis of synthesized materials [4]. |
| Robotic Electrochemical Workstation | Performs high-throughput performance testing (e.g., catalyst activity) without manual intervention [4]. |
| Computer Vision Monitoring System | Uses cameras and visual language models to monitor experiments in real-time, detecting issues and suggesting corrections to ensure reproducibility [4]. |
Workflow Visualization
The fundamental difference between the two methodologies is their structure: one is a linear, human-guided process, while the other is an iterative, AI-guided loop. The diagrams below illustrate these distinct workflows.
Traditional Catalyst Development Workflow
AI-Driven Catalyst Development Workflow
The data presented in this comparison leads to an unambiguous conclusion: AI-driven catalyst development represents a paradigm shift with superior metrics in both timelines and cost-effectiveness for complex discovery tasks. While traditional methods offer predictability and remain valuable for certain problems, their manual nature inherently limits the scale and speed of exploration. The AI-driven approach, characterized by its non-linear, active learning loop and robotic automation, demonstrates a capacity to explore vast chemical spaces more thoroughly and identify high-performing solutions orders of magnitude faster. The documented case of discovering a record-breaking fuel cell catalyst in three months, a task that could have taken years traditionally, serves as a powerful testament to this new capability [4]. For research organizations aiming to remain at the forefront of innovation in catalyst and drug development, the integration of AI-driven platforms is no longer a speculative advantage but a strategic necessity to accelerate time-to-discovery and maximize the return on research investment.
This guide provides an objective comparison of performance between traditional and AI-driven approaches in drug development, with a specific focus on candidate quality and subsequent clinical progression. The analysis is framed within a broader research thesis comparing traditional and AI-driven catalyst development.
The integration of Artificial Intelligence (AI) into drug development represents a paradigm shift from serendipity-driven discovery to engineered design. The following table summarizes the core performance differences between traditional and AI-driven methodologies, highlighting their impact on candidate quality and clinical success.
Table 1: Core Performance Metrics: Traditional vs. AI-Driven Drug Development
| Metric | Traditional Approach | AI-Driven Approach | Data Source / Context |
|---|---|---|---|
| Average Development Time | 10-15 years [58] [59] | Reduced by 1-4 years; Preclinical stage slashed from 5-6 years to 12-18 months [48] [59] | Industry-wide analysis |
| Average Cost per Drug | ~$2.6 billion [48] [58] | Preclinical costs reduced by 25-50% [58] | Boston Consulting Group, McKinsey |
| Clinical Trial Phase I Success Rate | 40-65% (industry average) [58] | 80-90% (for AI-discovered drugs) [58] | Analysis of AI-discovered drug pipelines |
| Probability of Clinical Success (All Phases) | ~10% [48] | Projected to increase by ~20% [60] | BiopharmaTrend, industry reports |
| Typical Discovery Method | High-throughput screening (trial-and-error) [59] | De novo molecular design & target identification [48] [58] | Company platforms (e.g., Insilico, Exscientia) |
| Impact of Candidate Quality | High late-stage attrition due to poor target or molecule selection [61] | Higher-quality candidates with designed properties enter trials, reducing late-stage failure risk [48] [59] | Analysis of clinical outcomes |
Clinical Progression: A Data-Driven Comparison
The ultimate validation of any drug discovery approach is its success in human clinical trials. The following table compares the clinical progression of selected AI-driven drug candidates against traditional industry benchmarks.
Table 2: Clinical Progression Benchmarks: Select AI Candidates vs. Traditional Norms
| Drug Candidate / Benchmark | Discovery Approach | Indication | Clinical Stage & Key Outcome | Comparative Performance |
|---|---|---|---|---|
| ISM001-055 (Insilico Medicine) | AI-designed novel target (TNIK) and novel molecule [59] | Idiopathic Pulmonary Fibrosis (IPF) [59] | Phase IIa: Demonstrated dose-dependent efficacy (98.4 mL FVC improvement vs. placebo decline) [59] | Target-to-Preclinical Candidate: 18 months (vs. multi-year average) [61] [59] |
| REC-994 (Recursion) | AI-repurposed existing molecule [59] | Cerebral Cavernous Malformation (CCM) [59] | Discontinued after long-term extension failed to show sustained efficacy [59] | Highlights the "translation gap"; AI can find activity but human biology complexity remains a challenge [59] |
| AI-Discovered Drug Average (Phase I) | Various AI platforms [58] | Various | Phase I Trials | 80-90% Success Rate (vs. 40-65% traditional average) [58] |
| Traditional Industry Benchmark | Traditional HTS & design [61] | Various | Phase II to Phase III Transition | High failure rate; ~90% attrition from clinical entry to approval [61] [58] |
Experimental Protocols: How AI Performance is Measured
To ensure a fair comparison, the performance of AI-driven methods is validated through rigorous, domain-specific experimental protocols. These methodologies underpin the data presented in the previous tables.
Protocol: AI-Driven Target Identification and Molecule Design
This protocol details the end-to-end process for discovering a novel target and designing a novel drug candidate, as exemplified by Insilico Medicine's ISM001-055 program [59].
- Objective: To identify a novel therapeutic target for a complex disease and design a novel, selective small molecule inhibitor de novo.
- 1. Target Identification (PandaOmics Platform):
- Input: Multi-modal biological data (genomics, proteomics, transcriptomics, literature) [59].
- AI Method: Deep learning models and natural language processing (NLP) analyze datasets to rank and identify novel disease-associated targets without prior clinical validation [58] [59].
- Output: A novel target hypothesis (e.g., TNIK for IPF) [59].
- 2. Molecular Design (Chemistry42 Platform):
- Input: The identified target and desired molecular properties (e.g., potency, selectivity, ADMET) [59].
- AI Method: Generative AI and reinforcement learning generate and iteratively optimize novel molecular structures that are predicted to meet the target product profile [48] [58].
- Output: A set of novel, AI-designed small molecule candidates for synthesis and testing [59].
- 3. Experimental Validation:
- Key Performance Metric: Time from program initiation to nomination of a Preclinical Candidate (PCC). Insilico Medicine reported 18 months for this process [61] [59].
Protocol: Catalyst Design Evaluation with CatScore
In the broader context of catalyst development for molecular synthesis, AI-driven evaluation methods provide a significant speed advantage over traditional computational chemistry. This protocol is critical for rapidly assessing the quality of catalysts designed for synthesizing drug intermediates and active pharmaceutical ingredients (APIs) [62].
- Objective: To rapidly and accurately evaluate the predicted selectivity and effectiveness of a novel catalyst design.
- 1. Model Training:
- Data: A dataset of experimentally validated reaction-catalyst-product tuples (e.g., the AHO dataset for asymmetric hydrogenation) [62].
- AI Model: A product prediction model (e.g., based on T5Chem/CodeT5 architecture) is trained to predict the final product distribution given reactants and a catalyst [62].
- 2. Evaluation (CatScore):
- Input: The reactants (r), target product (p), and the designed catalyst (d(r,p)).
- Process: The trained product prediction model (fθ) calculates the probability (qθ) it assigns to the target product being formed.
- Output: CatScore (qθ), a proxy for predicted catalyst selectivity. A higher score indicates a more effective catalyst [62].
- Key Performance Metric:
- Speed: Evaluation takes ~3 CPU seconds per catalyst, compared to ~75 CPU hours for a traditional Density Functional Theory (DFT) and Linear Free Energy Relationship (LFER) approach [62].
- Accuracy: Predictions show a Spearman's ρ = 0.84 correlation with experimental outcomes, outperforming DFT-based LFERs (ρ = 0.55) [62].
Protocol: AI-Optimized Clinical Trial Design and Patient Recruitment
This protocol outlines how AI improves the quality and success rate of clinical trials, directly impacting the clinical progression of drug candidates [48] [58].
- Objective: To optimize clinical trial protocol design and accelerate the recruitment of suitable patients.
- 1. Trial Design Optimization:
- Input: Historical clinical trial data, real-world evidence (RWE), and the drug's target product profile.
- AI Method: Machine learning models simulate thousands of trial scenarios to predict optimal endpoints, inclusion/exclusion criteria, and dosing regimens. This can cut trial duration by up to 10% [48].
- 2. Patient Recruitment and Stratification:
- Input: De-identified Electronic Health Records (EHRs), genetic data, and trial criteria.
- AI Method: NLP and ML models (e.g., TrialGPT) analyze patient records to identify eligible participants with high accuracy, predicting dropouts and ensuring diverse populations [48].
- Output: A list of pre-qualified, likely-to-respond patients for recruitment [48] [58].
- Key Performance Metric: Companies like Sanofi report reducing patient recruitment timelines "from months to minutes" using AI tools. AI-enabled site selection can improve identification of top-enrolling sites by 30-50% [58] [59].
Workflow Visualization: AI-Driven Catalyst and Drug Design
The following diagram illustrates the integrated, closed-loop workflow for AI-driven catalyst and drug candidate design, showing how data and AI models create a continuous cycle of improvement.
AI-Driven Design Workflow
Clinical Pathway Visualization: AI vs. Traditional
This diagram contrasts the clinical progression pathways and key decision points for candidates developed via traditional versus AI-driven methods.
Clinical Progression Pathway
The Scientist's Toolkit: Research Reagent Solutions
This table details key computational tools, platforms, and data resources that are essential for conducting AI-driven drug and catalyst development research.
Table 3: Essential Research Tools for AI-Driven Development
| Tool / Resource Name | Type | Primary Function | Relevance to Candidate Quality |
|---|---|---|---|
| AlphaFold (DeepMind) [48] | AI Model | Accurately predicts 3D protein structures from amino acid sequences. | Enables structure-based drug design against previously inaccessible targets. |
| Chemistry42 (Insilico) [59] | Software Platform | Generative AI engine for de novo design of novel small molecules. | Generates novel chemical entities with optimized properties for a specific target. |
| PandaOmics (Insilico) [59] | Software Platform | AI-powered multi-modal data analysis for novel target identification. | Identifies novel, druggable targets with high disease relevance, de-risking early discovery. |
| CatScore [62] | Evaluation Metric | A learning-based metric for rapid, accurate prediction of catalyst selectivity. | Accelerates and improves the design of catalysts for synthes drug intermediates. |
| ASKCOS [1] | Software Platform | Open-source platform for computer-aided synthesis planning (CASP). | Designs feasible synthetic routes for AI-designed molecules, bridging design and manufacture. |
| AHO Dataset [62] | Chemical Dataset | A curated resource for studying asymmetric hydrogenation of olefins. | Provides high-quality, experimental data for training and validating AI catalyst models. |
| Exscientia's Centaur Chemist [48] | Software Platform | AI-driven drug design platform that automates molecule design and optimization. | Reportedly designed a cancer drug candidate that entered trials in ~1 year. |
| Recursion's Phenomics [59] | Software Platform | Uses cellular imaging and AI to link drug-induced morphological changes to biology. | Generates novel biological insights for drug repurposing and target identification. |
The pharmaceutical industry is undergoing a profound transformation driven by artificial intelligence, with adoption rates surging across all sectors of healthcare. Recent data reveals that 22% of healthcare organizations have now implemented domain-specific AI tools, representing a 7x increase over 2024 and a 10x increase over 2023 [63]. Health systems lead this adoption at 27%, with pharmaceutical and biotechnology companies demonstrating significant momentum in deploying AI to accelerate drug development and optimize clinical trials [63]. This rapid uptake signals a major shift in how pharmaceutical research and development is conducted, moving AI from experimental pilots to core strategic capabilities.
Current Adoption Metrics and Market Trajectory
The AI revolution in pharmaceuticals is quantifiable through both spending figures and utilization rates. Understanding these metrics provides critical context for the industry's direction and investment priorities.
Table 1: AI Adoption Rates Across Healthcare Sectors (2025)
| Sector | Adoption Rate | Primary Focus Areas | Procurement Cycle Time |
|---|---|---|---|
| Health Systems | 27% | Workflow automation, clinical documentation | 6.6 months (reduced from 8.0) |
| Outpatient Providers | 18% | Patient engagement, administrative automation | 4.7 months (reduced from 6.0) |
| Payers | 14% | Prior authorization, claims processing | 11.3 months (increased from 9.4) |
| Pharma & Biotech | Earlier stage but accelerating | Drug discovery, clinical trial optimization | ~10 months (steady) |
| Broad Economy Average | 9% | General-purpose AI tools | Varies widely |
Source: Menlo Ventures 2025 Report [63]
The financial commitment to AI is equally substantial. Healthcare AI spending reached $1.4 billion in 2025, nearly tripling from the previous year [63]. Within this investment, 85% flows to startups rather than legacy incumbents, indicating significant market disruption [63]. The pharmaceutical AI market specifically is estimated at $1.94 billion in 2025 and is forecast to reach $16.49 billion by 2034, accelerating at a CAGR of 27% from 2025 to 2034 [48].
Table 2: Pharmaceutical AI Market Projections and Impact Metrics
| Metric | 2025 Status | Projected Future Value | Timeframe |
|---|---|---|---|
| Overall Pharma AI Market | $1.94 billion | $16.49 billion | 2034 |
| Annual Value Generation | - | $350-410 billion | Annual by 2025 |
| AI-Discovered New Drugs | - | 30% of all new drugs | 2025 |
| Drug Discovery Cost Reduction | - | 40% savings | Current potential |
| Clinical Trial Cost Savings | - | $25 billion | Industry-wide |
Sources: Coherent Solutions, Menlo Ventures, McKinsey Analysis [48] [63] [19]
Key Players and Strategic Approaches
Leading Pharmaceutical Companies
The industry's major players have embraced AI through distinct strategic pathways:
- Pfizer has partnered with Tempus, CytoReason, and Gero to integrate AI into drug discovery, clinical trials, and patient population analysis, notably accelerating COVID-19 treatments including Paxlovid [48].
- AstraZeneca collaborates with BenevolentAI and Qure.ai to develop treatments for chronic kidney disease and pulmonary fibrosis while optimizing clinical trial designs [48].
- Janssen (Johnson & Johnson) runs over 100 AI projects in clinical trials, patient recruitment, and drug discovery, utilizing their Trials360.ai platform to streamline processes [48].
- Roche tops the Statista AI readiness index, building capabilities through both in-house talent and strategic acquisitions of smaller, tech-driven firms [48].
"AI-First" Biotech Innovators
A new class of biotech companies has emerged with AI as their foundational capability:
- BenevolentAI specializes in AI-powered drug discovery and target selection, with partnerships including AstraZeneca and Novartis [48].
- Insilico Medicine utilizes deep learning models for drug design and synthesis, significantly accelerating discovery timelines [48].
- Unlearn employs digital twin technology to create AI-driven models of disease progression, reducing required clinical trial participants while maintaining statistical integrity [50].
Methodological Framework: Traditional vs. AI-Driven Catalyst Development
The transition from traditional to AI-driven approaches represents a fundamental shift in pharmaceutical research methodology, particularly in molecular catalysis and drug discovery.
Comparative Experimental Protocols
Table 3: Traditional vs. AI-Driven Catalyst Development Workflows
| Research Phase | Traditional Approach | AI-Driven Approach | Key Differentiators |
|---|---|---|---|
| Retrosynthetic Design | Manual literature search (Reaxys, SciFinder); expert intuition | AI-powered retrosynthesis tools (ASKCOS, AiZynthFinder); template-based analysis | Automated route generation; exploration beyond published reactions |
| Catalyst Design | Linear free energy relationships (LFERs); limited SAR datasets | AI exploration of high-dimensional chemical space; pattern recognition in complex datasets | Vastly expanded parameter optimization; novel catalyst identification |
| Reaction Optimization | One-variable-at-a-time experimentation; labor-intensive screening | Autonomous experimentation; robotic flow chemistry platforms | Dramatically reduced optimization time; continuous reaction improvement |
| Validation | Sequential peer review; limited dataset validation | Digital twin simulations; predictive outcome modeling | Pre-validation of hypotheses; reduced experimental failure rate |
Sources: Organic Chemistry Frontiers, SN Analytics, Drug Target Review [50] [64] [1]
Workflow Visualization
AI-Driven vs. Traditional Catalyst Development Workflows
Experimental Protocols and Validation Frameworks
Digital Twin Clinical Trial Methodology
The application of AI in clinical trials represents one of the most mature use cases, with validated methodologies emerging:
Protocol Objective: To reduce clinical trial size and duration while maintaining statistical power through AI-generated digital twins.
Experimental Design:
- Historical Data Collection: Aggregate comprehensive clinical data from previous trials, including patient demographics, disease progression metrics, and treatment outcomes.
- Model Training: Develop AI models that predict individual patient disease progression trajectories based on baseline characteristics.
- Digital Twin Generation: For each actual patient in the treatment arm, create a matched digital twin that simulates the patient's expected disease progression without intervention.
- Outcome Comparison: Compare actual treatment outcomes against digital twin predictions to determine drug efficacy.
- Statistical Validation: Implement guardrails to ensure Type 1 error rates remain controlled, addressing regulatory concerns [50].
Validation Metrics: Trial sponsors utilizing this approach have demonstrated the potential to reduce control arm sizes in Phase III trials significantly, with particular impact in costly therapeutic areas like Alzheimer's where patient costs can exceed $300,000 each [50].
AI-Driven Retrosynthesis Implementation
Protocol Objective: To accelerate molecular synthesis planning through AI-powered retrosynthetic analysis.
Experimental Workflow:
- Target Input: Define desired molecular structure with specified complexity parameters.
- Template Application: Employ AI systems (e.g., ASKCOS, AiZynthFinder) with automated template extraction from databases like Reaxys and USPTO containing 12.5+ million single-step reactions.
- Route Generation: Apply Monte Carlo tree search algorithms to identify optimal synthetic pathways.
- Experimental Validation: Execute predicted routes on robotic flow chemistry platforms for empirical verification.
- Iterative Refinement: Use experimental outcomes to refine AI prediction models [1].
Performance Metrics: This approach enabled the design and experimental validation of complete synthetic routes for complex natural products including (–)-Dauricine, Tacamonidine, and Lamellodysidine A, with Turing tests demonstrating that experienced chemists could not distinguish between AI-generated routes and literature-reported syntheses [1].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key AI and Experimental Platforms for Pharmaceutical Research
| Tool Category | Representative Solutions | Primary Function | Research Application |
|---|---|---|---|
| Retrosynthesis Platforms | ASKCOS, AiZynthFinder, Chemitica | Automated synthetic route design | Molecular synthesis planning |
| Clinical Trial AI | Unlearn's Digital Twin Generator | Predictive patient modeling | Clinical trial optimization |
| Drug Discovery AI | Insilico Medicine's Platform, BenevolentAI | Target identification & compound design | Novel therapeutic discovery |
| Data Analysis Environments | RDChiral, custom Python/R workflows | Chemical data processing & pattern recognition | Experimental data analysis |
| Automation Controllers | Robotic flow chemistry platforms | High-throughput experimentation | Reaction optimization & validation |
| Knowledge Management | Reaxys, SciFinder with AI augmentation | Literature-based discovery | Research context & precedent analysis |
Sources: Organic Chemistry Frontiers, SN Analytics, Industry Reports [48] [64] [1]
Future Trajectory and Emerging Capabilities
The adoption momentum shows no signs of slowing, with several transformative trends emerging:
AI Agent Proliferation: Organizations are increasingly experimenting with AI agents—systems capable of planning and executing multiple steps in workflows. Currently, 23% of organizations report scaling agentic AI systems, with an additional 39% experimenting [19]. In pharmaceutical contexts, these agents are appearing in IT management, knowledge discovery, and research automation.
Rare Disease Innovation: Improved data efficiency enables powerful AI models to work with smaller datasets, opening breakthroughs in rare diseases and niche cancer subtypes where data scarcity has traditionally limited research [50].
Generative AI Expansion: Beyond small molecules, generative AI is advancing into biologics design, with 75% of pharmaceutical companies making generative AI a strategic priority for 2025 [64].
Workflow Transformation: Successful organizations are fundamentally redesigning workflows around AI capabilities rather than simply automating existing processes. AI high performers are three times more likely to have redesigned individual workflows compared to peers [19].
The pharmaceutical industry's AI adoption landscape reveals an sector at a tipping point, transitioning from isolated experiments to comprehensive transformation. The companies realizing greatest value are those treating AI not as a tool but as a catalytic force that reshapes decision-making, processes, and ultimately, therapeutic innovation.
The development of catalysts has long been a cornerstone of chemical innovation, with profound implications for pharmaceutical synthesis, energy sustainability, and industrial manufacturing. Traditional catalyst development has predominantly operated through a trial-and-error methodology, guided by expert intuition and incremental improvements based on established reaction mechanisms. While this approach has yielded significant successes, it inherently limits the exploration of chemical space and constrains the novelty of discoverable catalysts. The emergence of artificial intelligence (AI) has fundamentally transformed this landscape, introducing not only unprecedented speed but qualitatively different approaches to innovation and candidate discovery.
This comparison guide objectively analyzes the performance of traditional versus AI-driven catalyst development approaches, with particular focus on their differential impacts on the novelty of generated catalysts and the fundamental nature of the innovation process. While quantitative metrics demonstrate clear advantages in efficiency, the more profound distinction lies in how these paradigms expand the boundaries of discoverable chemical space and redefine what constitutes a viable catalyst candidate. Through examination of experimental data, methodological frameworks, and case studies, this guide provides researchers with a comprehensive assessment of how AI technologies are reshaping catalyst discovery at a conceptual level.
Comparative Analysis: Traditional vs. AI-Driven Catalyst Development
Table 1: Performance Comparison of Traditional vs. AI-Driven Catalyst Development
| Performance Metric | Traditional Approach | AI-Driven Approach | Qualitative Implications |
|---|---|---|---|
| Exploration Efficiency | Sequential testing of limited candidate libraries | High-throughput screening of vast chemical spaces | AI enables exploration beyond human intuition and established chemical knowledge |
| Candidate Novelty | Incremental modifications of known scaffolds | De novo generation of unprecedented structures | AI discovers catalysts with novel architectures not previously considered |
| Data Utilization | Relies on limited, curated datasets | Learns from diverse, multi-modal data (computational, experimental) | Identifies complex, non-linear structure-activity relationships |
| Innovation Process | Hypothesis-driven, linear optimization | Data-driven, parallel exploration with closed-loop validation | Transforms research from incremental improvement to fundamental discovery |
| Descriptor Identification | Manual, based on established theoretical frameworks | Automated discovery of non-intuitive descriptors and "catalyst genes" | Reveals previously unrecognized structure-property relationships [65] |
Table 2: Quantitative Performance Metrics from Experimental Studies
| Experimental Measurement | Traditional Methods | AI-Driven Systems | Improvement Factor |
|---|---|---|---|
| Time to Catalyst Identification | Months to years | Days to weeks | 5-10x acceleration [2] |
| Chemical Space Exploration | 10²-10³ candidates | 10⁵-10⁶ candidates | 100-1000x expansion [66] |
| Prediction Accuracy (Yield) | Limited quantitative prediction | RMSE: 0.7-1.2 (normalized) | Competitive or superior to DFT [66] |
| Success Rate in Experimental Validation | 5-15% (based on historical data) | 20-35% (reported in case studies) | 2-3x improvement [1] |
| Multi-parameter Optimization | Typically <3 parameters simultaneously | 5-10 parameters simultaneously | Enables complex trade-off optimization |
Methodological Frameworks: Experimental Protocols and Workflows
Traditional Catalyst Development Workflow
Traditional catalyst development follows a sequential, hypothesis-driven workflow that relies heavily on domain expertise and established chemical principles. The process begins with extensive literature review and formulation of hypotheses based on existing mechanistic understanding. Researchers then design catalyst candidates typically through incremental modifications of known scaffolds—adjusting ligands, metal centers, or support materials based on previous successful systems. Synthesis and characterization follow, employing standard chemical techniques to produce and analyze the proposed catalysts. Performance testing evaluates key metrics such as activity, selectivity, and stability under relevant reaction conditions. Data analysis provides feedback to refine the initial hypothesis, creating an iterative cycle that gradually converges toward improved catalysts. This methodology, while systematic, inherently limits exploration to chemical spaces proximal to existing knowledge and is constrained by the throughput of synthetic and testing capabilities.
AI-Driven Catalyst Development Workflow
AI-driven catalyst development implements a parallel, data-driven workflow that fundamentally transforms the exploration process. The workflow begins with aggregation of multi-source data including computational chemistry results (DFT calculations), experimental literature, and high-throughput experimentation data. AI model training employs various architectures—from graph neural networks to transformer-based models—to learn complex structure-property relationships from this aggregated data. The trained models then generate novel catalyst candidates through either predictive screening of virtual libraries or de novo molecular generation. These candidates undergo priority ranking based on predicted performance metrics and synthetic accessibility. High-priority candidates proceed to automated synthesis and testing using robotic platforms, generating performance data that feeds back to refine the AI models. This closed-loop system enables continuous improvement and exploration of chemical spaces far beyond human intuition, systematically generating high-novelty candidates with optimized properties [2] [66].
The CatDRX Framework for Catalyst Discovery
The CatDRX framework represents a cutting-edge approach in AI-driven catalyst discovery, employing a reaction-conditioned variational autoencoder (VAE) architecture specifically designed for catalyst generation and performance prediction [66]. This framework processes both catalyst structures and reaction conditions as inputs, generating a joint representation in latent space that captures complex relationships between catalyst features, reaction environments, and performance outcomes. The model is pre-trained on diverse reaction databases such as the Open Reaction Database (ORD) and fine-tuned for specific downstream applications. The encoder module maps input catalysts and conditions into a probabilistic latent space, while the decoder reconstructs catalyst structures conditioned on specific reaction requirements. A key innovation is the simultaneous prediction of catalytic performance (yield and related properties) alongside catalyst generation, enabling direct optimization toward desired objectives. This integrated approach allows for exploration of catalyst candidates specifically tailored to reaction constraints, significantly expanding the novelty and applicability of discovered materials compared to traditional template-based methods.
Experimental Protocols and Validation
AI-Driven Catalyst Gene Discovery Protocol
The AI-driven discovery of "catalyst genes" employs subgroup discovery (SGD) methodology to identify fundamental material properties that correlate with enhanced catalytic performance [65]. This protocol begins with first-principles calculations (typically DFT) for a broad family of candidate materials, computing both simple features (easily obtainable properties) and complex indicators of catalytic performance. The SGD algorithm then identifies subgroups with outstanding characteristics by evaluating combinations of feature thresholds that optimize target properties. Experimental validation involves synthesizing predicted high-performance catalysts and evaluating them under standardized conditions, with performance metrics compared against both AI predictions and traditionally developed benchmarks. This approach has successfully identified previously unrecognized catalyst genes for CO₂ conversion, including specific combinations of elemental properties and surface characteristics that promote strong elongation of C-O bonds—a key indicator of activation [65].
Autonomous Discovery Workflow Protocol
Fully autonomous catalyst discovery systems implement closed-loop workflows that integrate AI planning with robotic experimentation [2]. The protocol initiates with human-defined objectives and constraints, followed by AI-generated experimental plans that optimize for both performance and information gain. Automated synthesis platforms execute these plans, employing techniques ranging from sol-gel methods to chemical vapor deposition based on material requirements. High-throughput characterization techniques (XRD, XPS, TEM) provide structural data, while parallelized reactor systems evaluate catalytic performance. AI models continuously update based on experimental outcomes, refining subsequent experimentation cycles. This protocol has demonstrated the ability to discover optimized catalyst formulations in timeframes reduced from years to days, with recent implementations successfully developing oxygen evolution catalysts from Martian meteorites—showcasing the novelty and adaptability of AI-driven discovery [2].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for AI-Driven Catalyst Development
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Open Reaction Database (ORD) | Provides diverse reaction data for model pre-training | Contains ~2 million reactions; enables transfer learning [66] |
| DFT Calculation Suites | Generates training data and validates candidate properties | VASP, Quantum ESPRESSO; computes adsorption energies, activation barriers |
| Reaction Descriptors (RXNFPs) | Encodes reaction information for machine learning | 256-bit molecular fingerprints; enables reaction similarity analysis [66] |
| Automated Synthesis Platforms | Executes high-throughput catalyst preparation | Enables rapid experimental validation of AI predictions [2] |
| Structural Featurization Tools | Represents catalysts for machine learning models | Includes ECFP4 fingerprints, graph representations, SMILES encoding |
| Subgroup Discovery Algorithms | Identifies significant feature combinations for optimization | Discovers "catalyst genes" – material properties correlating with performance [65] |
Critical Analysis: Innovation Quality and Candidate Novelty
The qualitative differences between traditional and AI-driven catalyst development extend beyond efficiency metrics to fundamentally distinct innovation patterns. Traditional methods excel at incremental optimization within understood design paradigms, producing catalysts with well-characterized properties and predictable behavior. This approach minimizes risk and aligns with established manufacturing capabilities, but inherently constrains novelty to variations of known systems.
In contrast, AI-driven approaches demonstrate emergent capabilities for disruptive innovation through several mechanisms. First, they enable identification of non-intuitive descriptors and "catalyst genes" that escape human recognition due to complex, multi-parameter interactions [65]. Second, generative models like CatDRX can create entirely novel molecular architectures conditioned on specific reaction requirements, exploring regions of chemical space without precedent in existing literature [66]. Third, the ability to simultaneously optimize multiple objectives (activity, selectivity, stability, cost) allows discovery of candidates with balanced property combinations that might be overlooked in sequential optimization.
Case studies demonstrate this novelty quantitatively. AI-generated catalysts frequently exhibit structural motifs and element combinations not represented in training data, with analysis showing 15-30% of high-performing candidates containing genuinely novel features compared to existing databases [66]. Furthermore, these systems have discovered catalyst compositions with unexpected resistance to poisoning or unusual temperature response profiles—properties not explicitly optimized for during training. This capacity for serendipitous discovery of beneficial secondary characteristics represents a qualitative advancement beyond targeted optimization.
The most profound impact, however, may be in how AI systems redefine the catalyst design process itself. By learning complex structure-property relationships directly from data rather than relying on simplified theoretical models, these systems can identify candidates that perform well for reasons not fully explained by current mechanistic understanding. This creates a new paradigm where catalyst discovery can precede complete theoretical comprehension, potentially accelerating the development of entirely new catalytic mechanisms and applications.
The comparison between traditional and AI-driven catalyst development reveals a fundamental transition in scientific approach—from incremental optimization within known boundaries to exploratory discovery across vastly expanded chemical spaces. While traditional methods retain value for specific optimization tasks and benefit from established theoretical foundations, AI-driven approaches offer qualitatively different innovation potential through their capacity to identify non-intuitive design rules, generate structurally novel candidates, and continuously refine discovery strategies based on experimental feedback.
The evidence indicates that the most significant impact of AI in catalyst development is not merely accelerated discovery, but the emergence of genuinely novel candidate materials with properties and performance characteristics unlikely to be identified through human intuition alone. As these technologies mature, their integration with automated experimentation and multi-scale modeling promises to further expand the boundaries of discoverable catalyst space, potentially enabling solutions to longstanding challenges in energy, sustainability, and pharmaceutical development that have resisted traditional approaches.
For researchers, this transition necessitates developing new interdisciplinary skills that bridge domain expertise in catalysis with computational literacy in AI methodologies. The most productive path forward likely involves hybrid approaches that leverage the pattern recognition and exploration capabilities of AI while incorporating human expertise for hypothesis generation, experimental design, and mechanistic interpretation—creating a synergistic relationship that maximizes both innovation quality and practical applicability.
Conclusion
The comparison unequivocally shows that AI-driven catalyst development is not merely an incremental improvement but a fundamental transformation of the research paradigm. While traditional methods anchored in deep expertise remain valuable, AI offers unparalleled advantages in speed, cost-efficiency, and the ability to navigate complex design spaces. The synthesis of insights from all four intents reveals that the most successful future lies in a hybrid, 'centaur' model—leveraging AI for rapid prediction and automation while harnessing human expertise for strategic oversight and interpreting complex results. For biomedical research, this synergy promises to drastically shorten the path from discovery to clinic, enabling more targeted therapies and personalized medicine. Future progress will hinge on developing more interpretable AI models, creating larger multi-modal databases, and establishing robust regulatory frameworks for AI-driven discoveries, ultimately paving the way for fully autonomous discovery platforms.
References
- 1. AI molecular catalysis: where are we now? [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d4qo02363c]
- 2. Artificial intelligence for catalyst design and synthesis [https://www.sciencedirect.com/science/article/abs/pii/S259023852500181X]
- 3. Smart Catalysts: How AI is Designing More Efficient ... [https://www.chemcopilot.com/blog/smart-catalysts-how-ai-is-designing-more-efficient-chemical-reactions]
- 4. AI system learns from many types of scientific information ... [https://news.mit.edu/2025/ai-system-learns-many-types-scientific-information-and-runs-experiments-discovering-new-materials-0925]
- 5. Reac-Discovery: an artificial intelligence–driven platform ... [https://www.nature.com/articles/s41467-025-64127-1]
- 6. Open Catalyst experiments 2024 (OCx24) [https://ai.meta.com/blog/open-catalyst-simulations-experiments/]
- 7. Catalyst performance and experimental validation of a ... [https://www.sciencedirect.com/science/article/abs/pii/S2213343717303536]
- 8. Artificial Intelligence Revolutionizes Catalyst Design and ... [https://deng.dicp.ac.cn/info/1027/2931.htm]
- 9. Catal-GPT: AI-driven directed efficient design framework for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12421571/]
- 10. Machine Learning Descriptors for Data‐Driven Catalysis Study [https://pmc.ncbi.nlm.nih.gov/articles/PMC10401178/]
- 11. The AI-Powered Pathway to Advanced Catalyst Development [https://datascience.uchicago.edu/insights/the-ai-powered-pathway-to-advanced-catalyst-development/]
- 12. Nature:生物催化发现的范式革命——“实验-数据-AI”闭环 [https://news.bioon.com/article/c9609012e10f.html]
- 13. Catalysis meets machine learning: a guide to data-driven ... [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc05274b?page=search]
- 14. Machine learning for catalysis: Bridging data-driven ... [https://www.sciencedirect.com/science/article/abs/pii/S2468519425005415]
- 15. 化工系王笑楠课题组在AI用于多相催化剂的发现及设计研究 ... [https://www.tsinghua.edu.cn/info/1175/121427.htm]
- 16. Data-driven de novo design of super-adhesive hydrogels [https://www.nature.com/articles/s41586-025-09269-4]
- 17. North America Catalyst Market Trends 2025 to 2033 [https://www.linkedin.com/pulse/north-america-catalyst-market-trends-2025-2033-market-tech-radar-5dref/]
- 18. AI-Driven Industry Growth Trends 2025 [https://www.mezzi.com/blog/ai-driven-industry-growth-trends-2025]
- 19. The State of AI: Global Survey 2025 [https://www.mckinsey.com/capabilities/quantumblack/our-insights/the-state-of-ai]
- 20. The integration of artificial intelligence and high-throughput ... [https://www.sciencedirect.com/science/article/abs/pii/S1004954125001831]
- 21. Catalyst Market Analysis, Size, and Forecast 2024-2028 [https://www.technavio.com/report/catalyst-market-industry-analysis]
- 22. Heterogeneous catalyst design by generative models [https://www.oaepublish.com/articles/jmi.2025.38]
- 23. Millions of new materials discovered with deep learning [https://deepmind.google/blog/millions-of-new-materials-discovered-with-deep-learning/]
- 24. a global close-loop feedback powered by autonomous AI ... [https://www.oaepublish.com/articles/aiagent.2025.02]
- 25. AI tackles the challenge of materials structure prediction [https://www.cam.ac.uk/research/news/ai-tackles-the-challenge-of-materials-structure-prediction]
- 26. Elements must meet minimum color contrast ratio thresholds [https://dequeuniversity.com/rules/axe/4.8/color-contrast]
- 27. AI vs Traditional Methods: A Comparative Analysis of Data ... [https://superagi.com/ai-vs-traditional-methods-a-comparative-analysis-of-data-driven-decision-making-in-2025/]
- 28. Unlocking catalytic longevity: a critical review of catalyst ... [https://pubs.rsc.org/en/content/articlehtml/2025/ya/d5ya00015g]
- 29. Recent Advances and Future Perspectives in Catalyst ... [https://www.mdpi.com/2071-1050/17/16/7370]
- 30. laboratories in China: an embodied intelligence-driven... Autonomous [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00072f]
- 31. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 32. Autonomous Scientific Workflows [https://www.emergentmind.com/topics/autonomous-scientific-workflows]
- 33. Artificial intelligence—the great job maker or taker? - C&EN [https://cen.acs.org/business/informatics/Artificial-intelligence-great-job-maker/103/i3]
- 34. Artificial Intelligence-Driven Innovations in Oncology Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12232943/]
- 35. 综述:领先的人工智能驱动药物发现平台:2025年的市场格局 ... [https://www.ebiotrade.com/newsf/2025-11/20251112002210649.htm]
- 36. 清华万引教授:万倍加速催化剂设计,AI突破DFT瓶颈! [https://finance.sina.com.cn/tech/csj/2025-10-11/doc-inftpeum0367254.shtml]
- 37. Accelerating the discovery of new cancer therapies using AI [https://www.uchicagomedicine.org/forefront/cancer-articles/2024/november/accelerating-the-discovery-of-new-cancer-therapies-using-ai]
- 38. The AI revolution: how multimodal intelligence will reshape ... [https://www.nature.com/articles/s44387-025-00044-4]
- 39. 杭州电子科技大学团队用“AI化学家团队”破解化学反应条件 ... [https://news.qq.com/rain/a/20251112A03O7R00]
- 40. 礼来部署全球制药业规模最大、算力最强AI 工厂加速药物研发 [https://blogs.nvidia.cn/blog/lilly-ai-factory-nvidia-blackwell-dgx-superpod/]
- 41. A review of machine learning methods for imbalanced data ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00270b]
- 42. Critical role of data quality in enabling AI in R&D [https://www.deloitte.com/uk/en/blogs/thoughts-from-the-centre/critical-role-of-data-quality-in-enabling-ai-in-r-d.html]
- 43. Understanding Black Box AI: Challenges and Solutions [https://www.ewsolutions.com/understanding-black-box-ai/]
- 44. Accelerating AI Interpretability [https://fas.org/publication/accelerating-ai-interpretability/]
- 45. Black Box Problem in AI [https://www.geeksforgeeks.org/artificial-intelligence/black-box-problem-in-ai/]
- 46. Mastering Explainable AI in 2025: A Beginner's Guide to ... [https://superagi.com/mastering-explainable-ai-in-2025-a-beginners-guide-to-transparent-and-interpretable-models/]
- 47. AI Black Box: What We're Still Getting Wrong about Trusting ... [https://hyperight.com/ai-black-box-what-were-still-getting-wrong-about-trusting-machine-learning-models/]
- 48. AI in Pharma and Biotech: Market Trends 2025 and Beyond [https://www.coherentsolutions.com/insights/artificial-intelligence-in-pharmaceuticals-and-biotechnology-current-trends-and-innovations]
- 49. The Challenge of AI Interpretability [https://www.deloitte.com/us/en/what-we-do/capabilities/applied-artificial-intelligence/articles/ai-interpretability-challenge.html]
- 50. How AI will reshape pharma in 2025 [https://www.drugtargetreview.com/article/154981/how-ai-will-reshape-pharma-by-2025/]
- 51. Recent developments in the use of machine learning ... [https://www.sciencedirect.com/science/article/pii/S1385894725016936]
- 52. Article Artificial-intelligence-assisted design principle for ... [https://www.sciencedirect.com/science/article/pii/S2666675825001146]
- 53. Third NFDI Berlin-Brandenburg network meeting: AI as an ... [https://events.hifis.net/event/2123/contributions/16745/]
- 54. AI Development Cost Guide 2025 – Budget & Pricing Tips [https://www.trootech.com/blog/ai-development-cost]
- 55. AI Development Cost: The Complete Breakdown 2025 [https://appinventiv.com/blog/ai-development-cost/]
- 56. AI Benchmarks 2025: Performance Metrics Show Record ... [https://www.sentisight.ai/ai-benchmarks-performance-soars-in-2025/]
- 57. 200+ AI Statistics & Trends for 2025: The Ultimate Roundup [https://www.fullview.io/blog/ai-statistics]
- 58. Artificial Intelligence in Drug Development 2025 [https://iicrs.com/blog/artificial-intelligence-in-drug-development-2025-iicrs/]
- 59. How AI Is Irrevocably Altering the DNA of Drug Development [https://www.drugpatentwatch.com/blog/how-ai-is-already-changing-drug-development/?srsltid=AfmBOopfaszJW1qhBz9XJEj0hh6-P4HtR8yEQIoSKm2oDQcZ_7LMUtZ8]
- 60. AI in Drug Discovery: Increasing Success Rates by 20% [https://www.pharmiweb.com/press-release/2025-02-21/ai-in-drug-discovery-increasing-success-rates-by-20]
- 61. Progress, Pitfalls, and Impact of AI‐Driven Clinical Trials [https://pmc.ncbi.nlm.nih.gov/articles/PMC11924158/]
- 62. CatScore: evaluating asymmetric catalyst design at high ... [https://pubs.rsc.org/en/content/articlehtml/2024/dd/d4dd00114a]
- 63. 2025: The State of AI in Healthcare [https://menlovc.com/perspective/2025-the-state-of-ai-in-healthcare/]
- 64. AI in Pharmaceutical Industry: 2025 Guide & Use Cases [https://sranalytics.io/blog/ai-in-pharmaceutical-industry/]
- 65. Artificial-intelligence-driven discovery of catalyst genes ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8776738/]
- 66. Reaction-conditioned generative model for catalyst design ... [https://www.nature.com/articles/s42004-025-01732-7]