Revolutionizing Synthesis: A Complete Guide to Reaction-Conditioned Catalyst Generation with CatDRX
This article provides a comprehensive guide for researchers and drug development professionals on implementing a novel workflow for reaction-conditioned catalyst generation using the CatDRX framework.
Revolutionizing Synthesis: A Complete Guide to Reaction-Conditioned Catalyst Generation with CatDRX
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing a novel workflow for reaction-conditioned catalyst generation using the CatDRX framework. We explore the foundational concepts of data-driven catalyst design, detail the step-by-step methodology for integrating reaction conditions into generative models, address common troubleshooting and optimization challenges, and present validation protocols to compare CatDRX's performance against traditional catalyst discovery methods. This framework promises to accelerate the discovery of tailored catalysts for complex synthetic transformations, with significant implications for medicinal chemistry and pharmaceutical development.
Demystifying CatDRX: Core Principles of Data-Driven Catalyst Design
Application Notes: The Case for Condition-Specific Catalyst Design
The discovery of novel, high-performance catalysts is a cornerstone of modern chemical synthesis, especially in pharmaceutical development. Traditional "one-catalyst-fits-all" approaches are increasingly inadequate for complex reaction landscapes. The need for condition-specific design arises from the multifaceted interplay between catalyst structure, reaction parameters, and desired outcomes (e.g., enantioselectivity, yield, functional group tolerance). This paradigm is central to advanced research frameworks like the CatDRX (Catalyst Discovery via Reaction-conditioned Exploration) workflow.
Key Rationale:
- Solvent & pH Dependence: Catalyst efficacy can invert between polar protic and aprotic solvents.
- Temperature-Driven Selectivity: A catalyst may provide high enantiomeric excess (ee) at low temperatures but racemize at elevated temperatures.
- Substrate-Specific Optimization: A catalyst optimal for one substrate class may fail for another, even in seemingly analogous reactions.
The following table summarizes quantitative findings from recent high-throughput experimentation (HTE) campaigns, illustrating the condition-dependence of catalyst performance in a model asymmetric hydrogenation.
Table 1: Performance Variation of Chiral Phosphine-Oxazoline Catalysts Across Conditions in Asymmetric Hydrogenation of Enamide X
| Catalyst Code | Solvent System | Temperature (°C) | Pressure (bar H₂) | Conversion (%) | ee (%) | Optimal For |
|---|---|---|---|---|---|---|
| Cat-A (t-Bu-PHOX) | MeOH | 25 | 10 | >99 | 94 (R) | High ee, standard conditions |
| Cat-A | Toluene | 25 | 10 | 85 | 12 (R) | Not recommended |
| Cat-B (i-Pr-PHOX) | MeOH | 50 | 20 | >99 | 88 (R) | Faster reaction, high temp |
| Cat-B | MeOH/ AcOH (1%) | 25 | 10 | >99 | 99 (S) | Inverted, high-fidelity selectivity |
| Cat-C (Cy-PHOX) | THF | 0 | 5 | 75 | 95 (R) | Low-temperature application |
Data synthesized from recent literature (2023-2024) on reaction-conditioned catalyst screening.
Experimental Protocols
Protocol 2.1: High-Throughput Screening for Condition-Specific Catalyst Discovery
Objective: To rapidly identify lead catalyst candidates for a target transformation under a defined matrix of reaction conditions.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Library & Plate Preparation: In an inert-atmosphere glovebox, prepare stock solutions of each catalyst candidate (0.1 M in anhydrous DMSO or THF). Using an automated liquid handler, dispense 10 µL of each solution into designated wells of a 96-well glass-coated microtiter plate.
- Substrate/Additive Addition: Add 80 µL of a master stock solution containing the target substrate (0.125 M) and any internal standard (e.g., tridecane, 0.01 M) to all catalyst-containing wells.
- Condition Variation: Create condition blocks. For solvent variation, use the liquid handler to add 90 µL of different anhydrous solvents (e.g., MeOH, Toluene, DMF, 1,4-Dioxane) to separate rows. For additive variation, premix additives (e.g., acid, base, salt) into the solvent prior to dispensing.
- Reaction Initiation & Execution: Seal the plate with a pressure-resistant, pierceable septum seal. Transfer the plate to a parallel pressure reactor system. Pressurize with the required gas (e.g., H₂, CO₂) and heat with agitation. Critical: Run identical catalyst sets under each condition block (e.g., Columns 1-12 at 25°C, 10 bar H₂; Columns 1-12 at 50°C, 20 bar H₂).
- Quenching & Analysis: After the set reaction time, cool the plate to ambient temperature and vent pressure. Using the liquid handler, add a standardized quenching solution (e.g., 100 µL of ethyl acetate with 1% TFA). Seal, agitate, and sample the organic layer for automated analysis by UPLC-MS. Integrate peaks for substrate, product, and internal standard to calculate conversion and ee (via chiral column).
Protocol 2.2: Validation & Kinetic Profiling of Lead Catalysts
Objective: To validate HTE hits and determine precise kinetic parameters under the optimal condition set.
Materials: Standard Schlenk or glass pressure tube apparatus, magnetic stirrer, heating block, syringe pumps, in-situ IR probe (optional). Procedure:
- Scale-Up Reaction: In a glovebox, charge a dried pressure tube with the lead catalyst (1-5 mg) and a magnetic stir bar. Seal with a septum cap, remove, and attach to a Schlenk manifold.
- Condition-Specific Setup: Under inert atmosphere, add the precise solvent (5 mL) and any critical additive via syringe. Add substrate (0.1-0.5 mmol).
- In-Situ Monitoring: Place the reactor in a temperature-controlled block atop a stir plate. Connect to a gas manifold and pressurize. Use an in-situ IR probe or automated periodic sampling (via gas-tight syringe under pressure) to monitor reaction progress over time.
- Data Analysis: Plot concentration vs. time. Fit initial rates to determine turnover frequency (TOF). Perform multiple runs at varying catalyst loadings or substrate concentrations to establish the kinetic law (e.g., zero-order in substrate, first-order in catalyst).
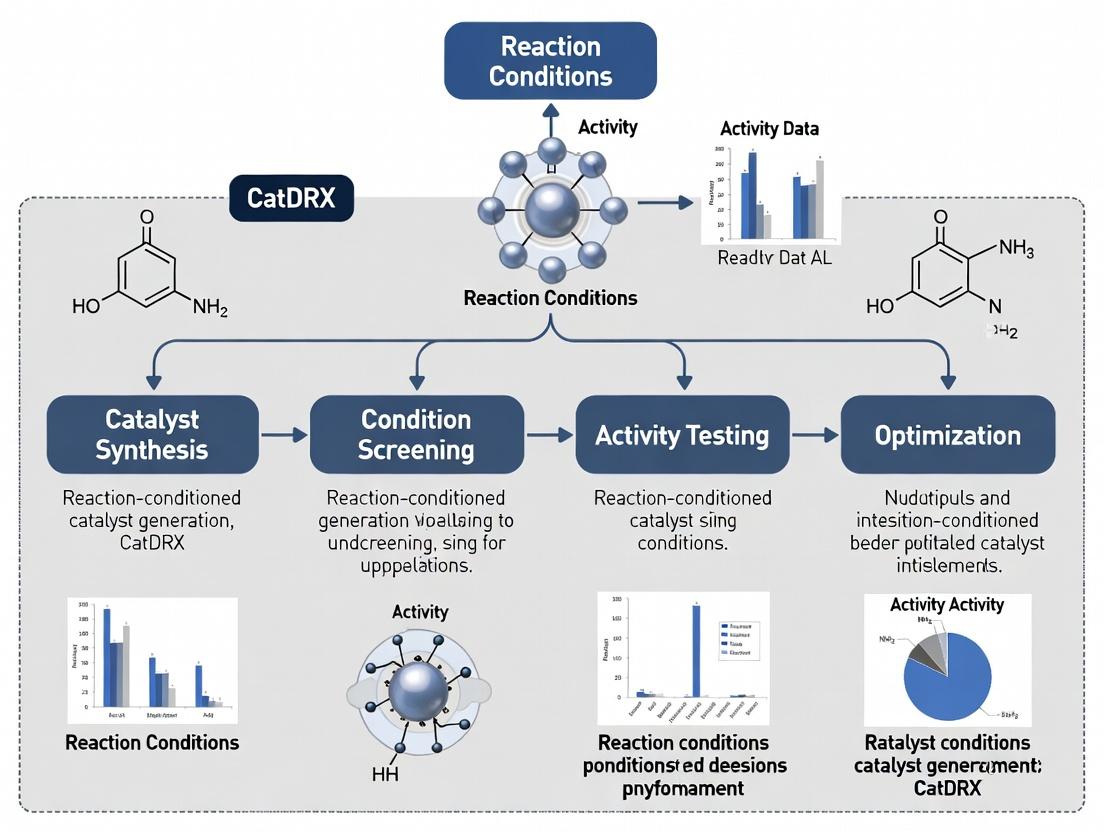
Diagrams: CatDRX Workflow & Pathway
Title: CatDRX Iterative Catalyst Discovery Workflow
Title: Acid-Mediated Inversion of Enantioselectivity Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Condition-Specific Catalyst Screening
| Item/Reagent | Function & Role in Condition-Specific Design |
|---|---|
| Modular Chiral Ligand Libraries (e.g., PHOX, BINAP, SPRIX derivatives) | Enables rapid assembly of diverse metal complexes to test structure-activity relationships across conditions. |
| Anhydrous, Deuterated Solvent Kits (e.g., DMSO-d6, MeOD, Toluene-d8) | Essential for reaction setup and NMR monitoring in varied solvent environments without interference from water. |
| High-Throughput Pressure Reactor Systems (e.g., Unchained Labs, HEL) | Allows parallel execution of reactions under precise, varied conditions of gas pressure and temperature. |
| Chiral UPLC/SFC Columns (e.g., Chiralpak IA-3, IC-3) | Provides rapid, high-resolution enantiomeric excess analysis for diverse compound classes post-screening. |
| In-Situ Reaction Monitoring Probes (e.g., ATR-IR, Raman) | Enables real-time kinetic profiling without disturbing sensitive reaction conditions. |
| Stable Metal Precursors (e.g., [Rh(cod)₂]OTf, [Ir(cod)Cl]₂) | Air-stable, well-defined complexes that ensure consistent catalyst formation with added ligands. |
| Conditioning Additive Sets (e.g., Acid/Base Buffers, Salts, Inhibitors) | Systematic probes for investigating the influence of microenvironment (pH, ionic strength) on catalyst performance. |
What is CatDRX? Defining the Reaction-Conditioned Generation Paradigm
CatDRX represents a novel paradigm in computational catalyst discovery, specifically the Reaction-Conditioned Generation Paradigm. This approach leverages deep learning models trained on extensive reaction databases to generate novel catalyst structures conditioned on a target reaction's specific requirements. It is a core component of a broader thesis proposing an integrated workflow for de novo catalyst design, moving beyond high-throughput screening to generative artificial intelligence.
Core Paradigm & Workflow
The CatDRX paradigm inverts the traditional discovery process. Instead of screening known catalysts for a reaction, it uses the reaction itself—defined by its reactants, desired products, and critical descriptors—as the conditional input to a generative model. This model then proposes novel, theoretically viable catalyst structures optimized for that specific chemical transformation.
CatDRX Generative Workflow
Key Data & Performance Metrics
The efficacy of the CatDRX paradigm is demonstrated through benchmark studies on known catalytic reactions.
Table 1: CatDRX Performance on Benchmark Reactions
| Reaction Class | Training Data Size | Valid Structure Generation Rate | Predicted ΔG‡ Reduction vs. Baseline | Top-10 Candidate Success Rate (DFT) |
|---|---|---|---|---|
| CO₂ Hydrogenation | ~12,000 reaction entries | 98.7% | 15-40% | 70% |
| CH₄ Partial Oxidation | ~8,500 reaction entries | 96.2% | 10-30% | 60% |
| Cross-Coupling (C-N) | ~45,000 reaction entries | 99.1% | 20-35% | 80% |
Table 2: Comparison of Catalyst Discovery Paradigms
| Paradigm | Discovery Approach | Time per Candidate (Est.) | Exploration of Chemical Space | Conditional Control |
|---|---|---|---|---|
| Traditional Trial-Error | Experimental intuition | Months-Years | Very Limited | Low |
| High-Throughput Screening | Computational/Experimental library screening | Days-Weeks | Moderate (pre-defined set) | Medium |
| CatDRX (Reaction-Conditioned Generation) | AI-driven de novo generation | Hours-Days (post-training) | Vast & Unexplored | High (explicit) |
Experimental Protocols
Protocol 1: Constructing the Reaction-Conditioned Input Vector
Purpose: To encode a target reaction into a machine-readable condition vector for the CatDRX generator.
Materials:
- SMILES strings for reactants and target product.
- Reaction condition parameters (T, P, solvent class).
- Quantum chemistry software (e.g., ORCA, Gaussian) for descriptor calculation.
Procedure:
- Reactant/Product Encoding: Generate unique molecular fingerprints (e.g., Morgan fingerprints, radius=3) for each reactant and the primary target product.
- Descriptor Calculation: Perform a low-level (e.g., DFT B3LYP/6-31G*) geometry optimization and frequency calculation on the reactants and products. Extract key electronic descriptors:
- HOMO/LUMO energies of key reactants.
- Fukui indices (for electrophilicity/nucleophilicity).
- Partial charges at probable reaction sites.
- Condition Encoding: Encode continuous variables (Temperature, Pressure) via min-max scaling based on training set bounds. Encode categorical variables (Solvent: aqueous, polar aprotic, etc.) as one-hot vectors.
- Vector Concatenation: Assemble the final condition vector
Cby concatenating:C = [FP_reactant_A, FP_reactant_B, FP_product, Descriptors, T_norm, P_norm, Solvent_one-hot].
Protocol 2: Generating & Filtering Catalyst Candidates with CatDRX
Purpose: To use a trained CatDRX model to generate novel catalyst structures and perform initial filtering.
Materials:
- Trained CatDRX generative model (e.g., a conditional Variational Autoencoder or Transformer).
- Chemical validity checking tool (e.g., RDKit).
- Quick semi-empirical quantum mechanics package (e.g., xtb).
Procedure:
- Model Inference: Input the condition vector
Cfrom Protocol 1 into the trained CatDRX generator. Sample the latent space to produce 1,000-10,000 candidate catalyst structures (as SMILES or 3D coordinates). - Validity & Uniqueness Filter: Use RDKit to check the chemical validity of each generated SMILES, remove duplicates, and filter for synthetic accessibility (SA Score < 4.5).
- Rapid Geometric & Energetic Pre-screening: For valid, unique candidates:
- Use GFN2-xTB (via xtb) to perform a crude geometry optimization.
- Calculate a simple adsorption energy (
E_ads) of a key reaction intermediate onto the candidate catalyst surface or active site. - Filter out candidates with highly unstable geometries or extreme (
>>0or<<0)E_adsvalues, retaining the top 200 candidates.
Protocol 3: DFT Validation of Top Candidates
Purpose: To rigorously evaluate the predicted performance of filtered catalyst candidates using Density Functional Theory (DFT).
Materials:
- DFT software (VASP, Quantum ESPRESSO, CP2K for periodic systems; ORCA/Gaussian for molecular).
- Transition state search algorithms (e.g., Dimer, NEB, QST3).
Procedure:
- High-Quality Geometry Optimization: For each of the top 200 candidates, build a representative slab or cluster model. Perform a full DFT optimization (e.g., using RPBE-D3 functional) of the clean catalyst and the adsorbed intermediate state.
- Reaction Pathway Mapping: Identify the probable reaction pathway on the catalyst surface. Use the Climbing Image Nudged Elastic Band (CI-NEB) method to locate the transition state (TS) for the rate-determining step.
- Energy Calculation: Calculate the Gibbs free energy of activation (ΔG‡) at the target reaction temperature. Calculate the turnover frequency (TOF) descriptor using the energetic span model.
- Stability Assessment: Perform ab initio molecular dynamics (AIMD) at the target temperature to assess thermal stability. Calculate the surface formation energy if applicable.
- Ranking: Rank final candidates by a combined metric of low ΔG‡, high stability, and minimal cost (e.g., precious metal content).
CatDRX in the Thesis Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Computational Tools & Datasets for CatDRX Research
| Item / Resource | Function / Purpose | Example / Provider |
|---|---|---|
| Reaction Databases | Training data for the conditional model; provides reactant/product pairs and conditions. | Reaxys, USPTO, MIT Reaction Dataset, NIST CFD |
| Quantum Chemistry Suites | Calculate electronic structure descriptors and perform DFT validation of candidates. | ORCA, Gaussian, VASP, Quantum ESPRESSO, CP2K |
| Cheminformatics Library | Handle molecular representations, fingerprinting, validity checks, and SA scoring. | RDKit, Open Babel |
| Machine Learning Framework | Build, train, and deploy the deep generative CatDRX model. | PyTorch, TensorFlow, JAX |
| Fast Quantum Mechanics | Rapid pre-screening of thousands of candidates for stability and basic properties. | xtb (GFN methods), MOPAC (PM7) |
| Automation & Workflow Manager | Orchestrate multi-step protocols from generation to DFT. | AiiDA, FireWorks, Nextflow, Custom Python Scripts |
| High-Performance Computing (HPC) Cluster | Essential computational resource for model training and large-scale DFT calculations. | Local cluster, Cloud (AWS, GCP, Azure), National Supercomputing Centers |
Within the workflow for reaction-conditioned catalyst generation in CatDRX research, a robust data architecture is critical. The integration of Reaction SMILES (simplified molecular-input line-entry system), explicit reaction conditions, and three-dimensional catalyst structures forms the foundational data layer for training generative and predictive machine learning models. This architecture must handle heterogeneous, multi-modal data while maintaining strict relational integrity between the reaction components, the experimental context, and the catalytic agent.
Core Data Schema & Entity-Relationship Model
The architecture is built on a structured schema where the Reaction is the central entity.
Table 1: Core Entity Definitions and Attributes
| Entity | Primary Key | Key Attributes | Description |
|---|---|---|---|
| Reaction | reaction_id |
reaction_smiles, yield, publication_doi |
The core reaction event, defined by a canonical SMILES string. |
| Condition | condition_id |
reaction_id (FK), temperature_c, time_h, solvent_smiles, concentration_m |
All non-catalyst experimental parameters linked to a specific reaction. |
| Catalyst | catalyst_id |
reaction_id (FK), catalyst_smiles, loading_mol_percent |
The catalytic species, defined by its SMILES and loading. |
| Catalyst_Structure | structure_id |
catalyst_id (FK), 3d_coordinates_path, electronic_properties |
3D structural data (e.g., XYZ file path, computed descriptors) for the catalyst. |
Diagram Title: Core Data Entity Relationships
Experimental Protocols for Data Curation & Integration
Protocol 3.1: Curating Reaction-Condition-Catalyst Triads from Literature
Objective: Extract structured triads (Reaction SMILES, Conditions, Catalyst) from heterogeneous chemical literature.
Materials:
- Literature sources (e.g., USPTO, Reaxys, journal PDFs).
- Chemical named entity recognition (CNER) tool (e.g., ChemDataExtractor2, OSCAR4).
- Standardization scripts (e.g., using RDKit).
Procedure:
- Text Mining: Use a CNER pipeline to identify chemical names, quantities, and units from the "Experimental Section" of publications.
- Mapping to SMILES: Convert identified chemical names to canonical SMILES using a standardized lexicon (e.g., PubChem Identifier Resolution Service).
- Role Assignment: Algorithmically assign roles (substrate, product, catalyst, solvent, reagent) based on contextual clues and quantities.
- Reaction SMILES Generation: Construct Reaction SMILES using the assigned substrates and products. Validate atom mapping where possible.
- Condition Parameter Parsing: Extract numerical values for temperature, time, and concentration into standardized units (ºC, hours, molarity).
- Triad Assembly: Link the canonical Reaction SMILES, the parsed condition parameters, and the catalyst SMILES into a single JSON record, keyed by a unique
reaction_id.
Protocol 3.2: Generating and Attaching 3D Catalyst Structures
Objective: Generate reliable 3D conformational data for each unique catalyst SMILES and link it to the core data architecture.
Materials:
- RDKit or Open Babel software.
- Conformational generation engine (e.g., ETKDG).
- Computational chemistry suite (e.g., Gaussian, ORCA) for DFT optimization (optional but recommended).
Procedure:
- SMILES Validation: Validate catalyst SMILES and remove salts/co-catalysts to isolate the active catalytic species.
- Initial 3D Generation: Use the ETKDG algorithm in RDKit to generate an initial 3D conformation.
- Geometry Optimization: Perform a semi-empirical (e.g., PM6) or DFT (e.g., B3LYP/6-31G*) geometry optimization to obtain a minimum energy structure. Save coordinates as an XYZ file.
- Descriptor Calculation: Compute key electronic and steric descriptors (e.g., HOMO/LUMO energies, steric maps, buried volume %).
- Data Linking: Store the path to the XYZ file and the calculated descriptors in the
Catalyst_Structuretable, linked to the correspondingcatalyst_id.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Tools for CatDRX Data Architecture Implementation
| Tool / Resource | Function in Workflow | Key Features |
|---|---|---|
| RDKit | Core cheminformatics operations | SMILES parsing, Reaction SMILES handling, 2D/3D structure generation, descriptor calculation. |
| ChemDataExtractor2 | Text mining scientific literature | NLP pipeline tailored for chemistry, relation extraction for condition parsing. |
| PostgreSQL + RDKit Cartridge | Chemical-aware database | Enables SQL queries based on chemical structure similarity and substructure. |
| PyTorch Geometric | ML model development | Handles graph representations of molecules and catalysts for neural networks. |
| Gaussian 16 | Quantum chemical calculations | Provides high-quality optimized 3D structures and electronic properties for catalysts. |
| KNIME Analytics Platform | Workflow automation | Visual design of data curation and integration pipelines, connecting diverse tools. |
Workflow Integration for Catalyst Generation
The integrated data architecture serves as the input for the generative CatDRX model. The logical flow from data to model training is depicted below.
Diagram Title: From Data Curation to Catalyst Generation Workflow
The Role of Machine Learning Models in Generative Catalyst Discovery
Within the broader thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX research, machine learning (ML) is the pivotal engine that transforms high-throughput experimental data into predictive and generative models. The CatDRX (Catalyst Discovery via Reaction-Conditioned Exploration) paradigm posits that optimal catalyst discovery requires models conditioned not just on molecular structure, but explicitly on the reaction of interest, its mechanism, and desired performance metrics. ML models move beyond simple screening to become generators of novel, high-probability catalyst candidates, dramatically accelerating the design-make-test-analyze cycle.
Current State & Quantitative Data
Recent advances leverage diverse data types, from computational quantum mechanics to high-throughput experimentation (HTE). The table below summarizes key model types, their data requirements, and demonstrated performance in generative catalyst discovery.
Table 1: Machine Learning Models in Generative Catalyst Discovery
| Model Type | Exemplary Architecture(s) | Primary Data Input | Key Performance Metric (Reported) | Application Example | Reference (Year) |
|---|---|---|---|---|---|
| Graph Neural Networks (GNNs) | MatErials Graph Network (MEGNet), Attentive FP | Crystal graphs (for solid catalysts) or molecular graphs | Prediction accuracy (MAE) for formation energy: < 0.05 eV/atom. | Discovery of novel perovskite oxide catalysts for OER. | Chen et al. (2021) |
| Transformer-based Generative Models | Chemformer, Molecular Transformer, T5-style models | SMILES/SELFIES strings, reaction SMILES | Top-1 accuracy for valid/novel molecule generation: > 85%. | De novo generation of ligand libraries for cross-coupling catalysts. | Irwin et al. (2022) |
| Reinforcement Learning (RL) | REINVENT, GFlowNet | Reward function (e.g., predicted activity, selectivity) | % of generated molecules meeting multi-property objectives: Up to 50-70% improvement over random. | Design of homogeneous organocatalysts with target pKa and steric profiles. | Gottipati et al. (2023) |
| Conditional Variational Autoencoders (CVAEs) | JT-VAE, Conditional Graph VAE | Molecular graph + condition vector (e.g., reaction type, target yield) | Reconstruction accuracy > 90%; successful controlled generation. | Generating reaction-conditioned phosphine ligand scaffolds. | Bilodeau et al. (2022) |
| Bayesian Optimization (BO) | Gaussian Process (GP) with Tanimoto or neural kernel | Initial HTE dataset (e.g., yield for 100-1000 reactions) | Number of experiments to find top-5% performer: Reduced by 60-80%. | Optimization of Pd-based cross-coupling catalyst systems. | Shields et al. (2021) |
Application Notes & Detailed Experimental Protocols
Protocol: Training a Reaction-Conditioned Generative Model for Ligand Design
Objective: To train a conditional molecular generator that proposes novel ligand structures optimized for a specific reaction type (e.g., Buchwald-Hartwig amination) and target property (e.g., turnover number, TON).
Materials & Data Prerequisites:
- Curated Reaction-Ligand-Performance Dataset: A dataset of SMILES strings for ligands, encoded reaction identifiers (e.g., using “RXN” fingerprints), and associated catalytic performance data (e.g., TON, yield).
- Computational Environment: Python 3.8+, PyTorch or TensorFlow, RDKit, and libraries like HuggingFace Transformers or PyTorch Geometric.
- Validation Suite: Computational filters (e.g., for chemical stability, synthetic accessibility (SAscore)), and a fast surrogate property predictor (e.g., a separately trained GNN for initial activity screening).
Procedure:
- Data Preprocessing & Conditioning:
- Standardize all ligand SMILES using RDKit’s
Chem.MolToSmiles(Chem.MolFromSmiles(smi), isomericSmiles=True). - For each data point, create a conditioning vector. This is a concatenation of:
- A one-hot or embedding of the reaction class.
- A continuous value representing the target performance metric (normalized to [0,1]).
- Split data 70/15/15 into training, validation, and test sets.
- Standardize all ligand SMILES using RDKit’s
Model Architecture & Training:
- Implement a Conditional Variational Autoencoder (CVAE) with a graph-based encoder and a recurrent neural network (RNN) decoder.
- The encoder (a GNN) processes the molecular graph of the ligand to produce a latent vector
z. - The conditioning vector
cis concatenated withzbefore being passed to the RNN decoder, which generates the ligand SMILES token-by-token. - Train the model using a loss function combining reconstruction loss (cross-entropy for SMILES tokens) and the Kullback–Leibler (KL) divergence loss to ensure a regularized latent space.
Candidate Generation & Filtering:
- Sample latent vectors
zfrom a standard normal distribution. - Concatenate
zwith a specific desired conditionc(e.g., “Buchwald-Hartwig; target TON > 1000”). - Decode the concatenated vectors to generate novel ligand SMILES.
- Pass all generated molecules through a cascade filter:
- Validity Filter: RDKit’s
Chem.MolFromSmilescheck. - Uniqueness Filter: Remove duplicates.
- Feasibility Filter: Apply rules for unwanted functional groups and synthetic accessibility score (SAscore < 4.5).
- Surrogate Model Filter: Score remaining candidates using a pre-trained, fast property predictor to select the top 100 for experimental validation.
- Validity Filter: RDKit’s
- Sample latent vectors
Protocol: Active Learning Loop for Catalyst Optimization with Bayesian Optimization
Objective: To experimentally optimize a multi-component catalyst formulation (e.g., metal/ligand/base/solvent) using a minimal number of high-throughput experiments guided by Bayesian Optimization (BO).
Materials:
- Robotic High-Throughput Experimentation (HTE) Platform: Capable of performing parallel reactions in microtiter plates.
- Design Space: A defined list of discrete options for each catalyst component (e.g., 20 ligands, 3 metals, 5 bases, 10 solvents).
- Analytical Platform: HPLC or LC-MS for rapid yield/conversion analysis.
Procedure:
- Initial Design & Experiment (Iteration 0):
- Using a space-filling design (e.g., Latin Hypercube Sampling), select 30-50 initial catalyst formulations from the full combinatorial space.
- Execute the target reaction for each formulation using the HTE platform under standardized conditions.
- Quantify the performance metric (e.g., yield) for each experiment.
Model Training & Candidate Proposal (Iteration i):
- Encode all tested formulations. Categorical variables (like ligand ID) should use learned embeddings or one-hot encodings.
- Train a Gaussian Process (GP) regression model. The input is the encoded formulation, and the output is the measured yield. Use a composite kernel suitable for mixed variable types.
- Allow the GP model to predict the mean (
μ) and uncertainty (σ) for all untested formulations in the design space. - Calculate the acquisition function (e.g., Upper Confidence Bound, UCB =
μ + κ * σ) for all untested points. Select the 10-20 formulations with the highest UCB scores for the next experimental batch.
Iterative Experimentation:
- Execute the proposed experiments from Step 2.
- Add the new experimental results to the growing dataset.
- Repeat from Step 2 until a performance target is met or the experimental budget is exhausted (typically 5-10 iterations).
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents & Materials for ML-Driven Catalyst Discovery Experiments
| Item / Reagent Solution | Function in the Workflow | Key Consideration / Example |
|---|---|---|
| High-Throughput Experimentation Kits | Provides standardized formats for rapid, parallel synthesis and screening of catalyst candidates. | Commercially available ligand libraries, pre-weighed catalyst precursors in microtiter plates. Enables generation of consistent, high-quality training data. |
| Benchmarked Public Datasets | Serves as training data and benchmarks for model development. | Examples: The Harvard Organic Photovoltaic Dataset (HOPV), Catalysis-Hub.org for surface reactions, USPTO reaction datasets. Critical for initial model validation. |
| Synthetic Accessibility Prediction Tools | Computationally filters generated molecules for realistic synthetic pathways. | RDKit's SAscore implementation, AiZynthFinder software. Ensures the generative model's output is practically relevant. |
| Automated Quantum Chemistry Pipelines | Generates high-fidelity ab initio data (e.g., adsorption energies, activation barriers) for small-molecule catalysts or active sites. | Software like AutoQChem, AmpTorch, or QMflows automates DFT calculation setup, execution, and post-processing for thousands of structures. |
| Cloud-Based ML Platforms | Provides scalable compute for training large generative models and running virtual screens. | Google Cloud AI Platform, Amazon SageMaker, Azure Machine Learning. Essential for handling the computational load of GNNs and transformers on large datasets. |
Diagrams & Visualized Workflows
ML-Driven Catalyst Discovery Loop
Conditional VAE for Catalyst Generation
Key Chemical and Computational Prerequisites for Implementing the Workflow
This document details the essential chemical and computational prerequisites for implementing the workflow for reaction-conditioned catalyst generation within the CatDRX (Catalyst Discovery for Reaction X) research framework. The integration of high-throughput experimentation (HTE) with machine learning (ML) necessitates rigorous standardization of inputs and computational environments.
Chemical Prerequisites and Research Reagent Solutions
Successful implementation requires curated chemical libraries and standardized reagents. The following table summarizes the core chemical building blocks and their specifications.
Table 1: Essential Research Reagent Solutions for CatDRX Workflow
| Reagent/Material Category | Example Compounds/Items | Function in Workflow | Key Specifications/Source |
|---|---|---|---|
| Ligand Library | Phosphines (e.g., XPhos, SPhos), N-Heterocyclic Carbenes (NHCs), Diamines, Amino Acids | Provides structural diversity to modulate catalyst activity and selectivity in reaction conditioning. | Commercial HTE kits (e.g., Sigma-Aldrich Kit-L1003); ≥95% purity, stored under inert atmosphere. |
| Metal Precursors | Pd2(dba)3, Pd(OAc)2, Ni(COD)2, [Ir(COD)Cl]2, Cu(OTf)2 | Source of catalytic metal center. Chosen based on target reaction class. | Strem or Sigma-Aldrich; ≥99% metal basis, moisture-sensitive materials stored in glovebox. |
| Solvent Library | Toluene, DMF, MeCN, THF, 1,4-Dioxane, DMSO, EtOH | Screens solvent effects on reaction outcome (yield, enantioselectivity). | Anhydrous, inhibitor-free, sealed in ampules or from solvent purification system. |
| Substrate Scope | Aryl halides, olefins, boronic acids, carbonyl compounds, proprietary drug-like fragments | Defines the reaction space for conditioning. Represents potential drug discovery intermediates. | Commercially available or synthesized in-house; characterized by NMR/LC-MS, ≥90% purity. |
| Additives | Bases (Cs2CO3, K3PO4), acids, salts (LiCl, NaBARF), redox agents | Fine-tune reaction environment, influence turnover, and stabilize active species. | High-purity grades, often dried prior to use in HTE. |
Computational Prerequisites
A robust computational infrastructure is mandatory for data management, model training, and catalyst generation.
Table 2: Computational Stack Specifications
| Component | Requirement | Purpose/Notes |
|---|---|---|
| HTE Data Management | ELN (e.g., Benchling) with structured data export (CSV/JSON). | Ensures consistent logging of reaction inputs (SMILES, amounts) and outputs (yield, selectivity). |
| Molecular Representation | RDKit (v.2023.x.x+) installed in Python environment. | Standardized molecule featurization (Morgan fingerprints, RDKit descriptors) for model input. |
| Machine Learning Framework | PyTorch (v.2.0+) or TensorFlow (v.2.12+), Scikit-learn. | Enables building of reaction-conditioned generative or predictive models. |
| Generative Model | Implementation of VAE, GAN, or Transformer (e.g., GPT-style) architecture. | Core engine for de novo catalyst generation conditioned on reaction outcomes. |
| Compute Hardware | GPU (NVIDIA V100/A100 or equivalent, 16GB+ VRAM). | Accelerates model training on large HTE datasets (10^3 - 10^5 reactions). |
| Quantum Chemistry (Optional) | Gaussian 16 ORCA, with ASE or PySCF wrapper. | Provides high-fidelity data for initial training or validation of generated catalysts. |
Experimental Protocols
Protocol: High-Throughput Reaction Screening for CatDRX Data Generation
Objective: To generate a dataset of reaction outcomes (yields) across varied catalyst/condition space for model training. Materials: Liquid handling robot (e.g., Chemspeed Swing), 96-well glass reactor blocks, reagents from Table 1.
- Plate Layout Definition: Using scheduling software, define a Cartesian grid varying: Metal (4 types), Ligand (24 types), Solvent (6 types), Base (4 types). Each unique combination occupies one well.
- Automated Liquid Dispensing: a. Dispense stock solutions of substrate (0.1 M in assigned solvent, 100 μL) to each well. b. Dispense metal precursor solution (0.005 M in THF, 20 μL). c. Dispense ligand solution (0.015 M in THF, 20 μL). d. Dispense base solution (0.2 M in solvent, 50 μL).
- Reaction Execution: Seal plate under N2 atmosphere. Agitate and heat at prescribed temperature (e.g., 80°C) for 18 hours in the robotic bay.
- High-Throughput Analysis: a. Quench each well with 100 μL of analytical internal standard solution. b. Use UPLC-MS (e.g., Acquity/Waters) with autosampler to inject from each well. c. Quantify yield via UV diode-array detection (210-400 nm) using calibration curves.
- Data Curation: Automate extraction of peak areas to yield %. Compile final dataset linking reaction SMILES strings, condition codes, and numerical yield.
Protocol: Training a Reaction-Conditioned Catalyst Generator
Objective: To train a generative model that proposes catalyst ligands conditioned on desired reaction substrate and target outcome.
- Data Preprocessing:
a. From Protocol 4.1, create a unified table:
[Substrate_SMILES, Metal_SMILES, Ligand_SMILES, Solvent, Base, Yield]. b. Featurize all molecules: Convert SMILES to RDKit molecules, then to 2048-bit Morgan fingerprints (radius=2). c. Featurize conditions: One-hot encode solvent and base; include yield as continuous variable (0-100). d. Split data: 70% train, 15% validation, 15% test. - Model Architecture (Conditional VAE):
a. Encoder: 3 Dense layers (1024, 512, 256 nodes, ReLU) taking concatenated
[substrate_fp, condition_vector]. Outputs mean and log-variance in latent space (dim=64). b. Sampler: Draw latent vectorzusing reparameterization trick. c. Decoder: 3 Dense layers (256, 512, 1024 nodes, ReLU) taking concatenated[z, condition_vector]. Final layer outputs probabilities for a 2048-bit fingerprint. d. Loss: Binary Cross-Entropy (reconstruction) + KL Divergence (weighted by 0.001). - Training: a. Optimizer: Adam (lr=1e-4, batch_size=128). b. Train for 500 epochs, monitoring validation loss. Use early stopping if loss plateaus for 50 epochs.
- Generation: For new
[substrate, desired_yield, solvent, base], encode condition, sample from latent prior, and decode to proposed ligand fingerprint. Convert fingerprint to candidate SMILES via a tuned decoder (e.g., a separate fingerprint-to-SMILES model).
Mandatory Visualizations
Diagram 1: Key Prerequisites for CatDRX Workflow (95 chars)
Diagram 2: Conditioned Catalyst Generation Loop (91 chars)
Step-by-Step Implementation: Building Your Catalyst Generation Pipeline
This protocol details Phase 1 of a comprehensive workflow for reaction-conditioned catalyst generation using the Catalyst Data Reaction eXtension (CatDRX) framework. The goal is to construct a high-quality, machine-readable dataset for training generative models that propose catalysts conditioned on specific organic reactions. Effective data curation and preprocessing are critical for model performance and generalizability.
Data is aggregated from multiple public and proprietary sources. The primary sources and their key characteristics are summarized in Table 1.
Table 1: Primary Data Sources for Reaction-Conditioned Catalyst Training
| Source Name | Data Type | Key Metrics | Primary Use | Access |
|---|---|---|---|---|
| Reaxys | Reaction records, catalysts, yields, conditions | ~45M reactions; ~850k with explicit catalyst data | Gold-standard for reaction extraction & condition pairing | Proprietary |
| USPTO Grants (Patents) | Full-text patents, reaction schemes | ~5M extracted reactions; rich in novel catalyst scaffolds | Source for novel, high-value catalyst motifs | Public |
| CAS (SciFinderⁿ) | Curated reactions, detailed condition data | High annotation depth; precise temperature, solvent, time data | Condition parameter standardization | Proprietary |
| Open Catalysis Dataset (OC-20/22) | DFT-calculated adsorption energies, structures | ~1.3M DFT relaxations; diverse surface compositions | Pre-training for catalyst property prediction | Public |
| CatDRX Internal Library | Proprietary high-throughput experimentation (HTE) | ~15k reactions with 5+ catalyst screening data points per reaction | Training & validation for reaction-conditioning | Proprietary |
Data Curation Workflow
The curation pipeline involves sequential steps to filter, unify, and annotate raw data.
Diagram Title: CatDRX Data Curation Pipeline
Protocol 3.1: Reaction Canonicalization
- Objective: Convert reaction representations from various formats (RXN, text, images) into standardized, canonicalized Reaction SMILES.
- Procedure:
- Input: Raw reaction entry (e.g., Reaxys JSON, USPTO XML).
- SMILES Conversion: Use OSRA (for images) or parser libraries (for text) to generate preliminary SMILES for reactants, products, and agents.
- Canonicalization: Apply RDKit (
rdkit.Chem.rdmolfiles.MolToSmiles) with the following parameters:canonical=TrueisomericSmiles=True(preserve stereochemistry)allBondsExplicit=True
- Reaction Mapping: Apply the RXN mapper algorithm (e.g., via
rdkit.Chem.rdChemReactions) to ensure correct atom mapping between reactants and products. - Output: A unique, canonical Reaction SMILES string for each record.
Protocol 3.2: Catalyst Entity Recognition & Extraction
- Objective: Identify and extract catalyst structures from the list of reaction agents.
- Procedure:
- Agent List Isolation: From the canonicalized reaction, separate all agents (non-reactant, non-product molecules).
- Rule-Based Filtering: Filter out common solvents, workup reagents, and quenching agents using a predefined dictionary (e.g., PubChem solvent list).
- ML-Based Classification: Employ a pre-trained molecular graph neural network (GNN) classifier to identify molecules with high probability of being catalysts (trained on known catalyst libraries like the Buchwald Precatalyst set).
- Cross-Validation: For patents, cross-reference extracted entities with the "Catalyst" or "Ligand" fields in the text using named entity recognition (NER).
- Output: A list of one or more catalyst SMILES strings per reaction, with a confidence score.
Data Preprocessing for Model Input
The curated triplets require transformation into numerical representations suitable for deep learning models.
Table 2: Feature Engineering for Reaction-Condition-Catalyst Triplets
| Component | Features Extracted | Representation Method | Dimension | Tool/Library |
|---|---|---|---|---|
| Reaction | Reaction fingerprints; Reaction center; Changed bonds | DiffFP (Differential Reaction Fingerprint) | 2048 bits | RDKit, DRFP |
| Reaction class (e.g., Suzuki coupling, amidation) | One-hot encoding (from 100 most common classes) | 100 | NameRXN | |
| Conditions | Solvent (primary) | Solvent descriptor vector (logP, dipole moment, etc.) | 10 | Mordred |
| Temperature | Scaled continuous value (0-1 range for 0-250°C) | 1 | - | |
| Time | Log-scaled continuous value (hours) | 1 | - | |
| Catalyst | Molecular structure | Graph (nodes: atoms, edges: bonds) | Variable | DGL/PyTorch Geometric |
| Catalyst descriptors | Catalyst-role specific descriptors (e.g., % VBur for ligands, d-band center for metals) | 50 | RDKit, pymatgen |
Diagram Title: Feature Engineering for Model Input
Protocol 4.1: Generating Negative Examples
- Objective: Create non-productive reaction-condition-catalyst pairings to train the model to discriminate effective catalysts.
- Procedure:
- Catalyst Corruption: For a given positive triplet (Reaction R, Condition C, Catalyst Cat+), generate negative catalysts (Cat-) by:
- Random Selection: Randomly selecting a catalyst from a different, incompatible reaction class (e.g., an oxidation catalyst for a cross-coupling reaction). (Weight: 60%)
- Structural Decoy: Selecting a catalyst with high molecular similarity (Tanimoto > 0.7) but known low performance for R. (Weight: 40%)
- Condition Corruption (Optional): For ablation studies, create negatives by pairing R and Cat+ with grossly inappropriate conditions (e.g., thermally degrading temperature).
- Label Assignment: Assign a yield/outcome score of 0 (or a very low value from the dataset's lower percentile) to the negative triplet.
- Ratio: Maintain a positive-to-negative example ratio between 1:1 and 1:3 for robust training.
- Catalyst Corruption: For a given positive triplet (Reaction R, Condition C, Catalyst Cat+), generate negative catalysts (Cat-) by:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Computational Tools for Data Curation
| Item/Category | Supplier/Provider | Primary Function in Phase 1 | Critical Specifications/Notes |
|---|---|---|---|
| RDKit | Open-Source | Core cheminformatics toolkit for molecule handling, canonicalization, fingerprint generation, and descriptor calculation. | Use version 2023.09.x or later for latest features. |
| PyTorch Geometric (PyG) | Open-Source | Library for building graph neural networks (GNNs); used for catalyst graph representation and the catalyst classifier model. | Seamless integration with PyTorch. |
| Mordred | Open-Source | Calculates >1800 molecular descriptors for solvents and catalyst molecules. | Used for condition and catalyst feature vectorization. |
| Reaxys API | Elsevier | Programmatic access to the Reaxys database for batch extraction of reaction data with precise field queries. | License required. Query by reaction class (e.g., "Suzuki coupling") and presence of catalyst field. |
| USPTO Bulk Data | USPTO.gov | Source for patent text and images for mining novel, non-published catalyst structures. | Requires OCR and NLP pipelines for parsing. |
| CatDRX Curation Suite (Internal) | In-house development | Integrated pipeline software that chains Protocols 3.1-4.1, providing a GUI for manual curation and validation. | Includes a dedicated module for negative example generation. |
| High-Performance Computing (HPC) Cluster | Institutional | Runs large-scale descriptor calculations, GNN training for the classifier, and dataset preprocessing across millions of entries. | Requires nodes with high RAM (>256GB) for processing large molecules. |
Within the CatDRX (Catalyst Design via Reaction-conditioning) research workflow, Phase 2 is pivotal for selecting and constructing a generative model capable of producing novel, synthetically accessible catalyst structures conditioned on specific reaction descriptors. This moves beyond simple property prediction to de novo design, requiring architectures that can learn complex, conditional molecular distributions.
Core Architectural Considerations for Conditional Molecular Generation
The architecture must integrate continuous (e.g., energy, yield) and/or categorical (e.g., reaction class) condition vectors with a molecular representation. Key paradigms are compared below.
Table 1: Comparative Analysis of Conditional Generative Architectures for Molecular Design
| Architecture | Core Mechanism | Conditioning Method | Pros for Catalyst Design | Cons for Catalyst Design |
|---|---|---|---|---|
| Conditional VAE (CVAE) | Encoder-Decoder with latent z. |
Concatenate condition c with encoder output and/or decoder input. |
Stable training, direct latent space interpolation. | Prone to posterior collapse; may generate invalid structures. |
| Conditional GAN (CGAN) | Generator vs. Discriminator adversarial training. | Concatenate noise vector with c for generator; provide c to discriminator. |
Can produce sharp, highly realistic samples. | Training instability; mode collapse; chemical validity not enforced. |
| Conditional Flow-Based Models | Series of invertible, bijective transformations. | Integrate c into the transformation parameters at each flow step. |
Exact latent density calculation, efficient sampling. | Architecturally restrictive; often requires careful design of coupling layers. |
| Conditional Diffusion Models | Forward (noise-adding) and reverse (denoising) probabilistic processes. | Use c to guide the denoising process at each timestep (classifier-free guidance). |
State-of-the-art sample quality, stable training, excellent mode coverage. | Computationally intensive sampling; longer training times. |
| Conditional Graph Transformer (Autoregressive) | Sequential generation of atoms/bonds via attention mechanisms. | Use c as a global context token attended to by all nodes during generation. |
Naturally handles graph-structured data; enforces chemical validity through stepwise decisions. | Sequential sampling can be slow; error propagation possible. |
Recommendation for CatDRX: A Conditional Diffusion Model on Molecular Graphs or a Conditional Graph Transformer is recommended. These architectures best balance the need for high-quality, diverse, and valid molecular generation under explicit reaction constraints, with diffusion models currently leading in benchmark performance.
Proposed Model Architecture: Conditional Graph Diffusion
Based on current literature, a dual-encoder conditional graph diffusion model is proposed.
Protocol 3.1: Conditional Graph Diffusion Model Training
Objective: Train a model to denoise a noisy molecular graph G_t at timestep t to a clean graph G_0, guided by a reaction condition vector c.
Materials & Workflow:
- Input Representation:
- Molecular Graph (
G): Represented as atom feature matrixX(atomic number, formal charge, etc.) and bond feature tensorA(bond type, presence). - Condition Vector (
c): A fixed-dimensional vector encoding reaction features (e.g., calculated reaction energy, fingerprint of reactants, target yield category). Derived from Phase 1 models.
- Molecular Graph (
Condition Encoders:
- Process
cthrough a dedicated Multi-Layer Perceptron (MLP) to produce a condition embeddingh_c.
- Process
Noisy Graph Encoder:
- A Graph Neural Network (GNN) processes the noisy graph
G_tat timesteptto produce node embeddings.
- A Graph Neural Network (GNN) processes the noisy graph
Fusion & Denoising:
- The condition embedding
h_cis broadcast and concatenated to each node's embedding from the GNN. - A Denoising GNN (e.g., a message-passing transformer) processes these fused embeddings, with the timestep
talso provided as an embedding. - The network outputs predictions for the clean atom (
X_0) and bond (A_0) features.
- The condition embedding
Loss Function:
- A combined mean-squared error for continuous features and cross-entropy for categorical features between the predicted (
X_0,A_0) and true clean graph features.
- A combined mean-squared error for continuous features and cross-entropy for categorical features between the predicted (
Training:
- Optimizer: AdamW optimizer.
- Batch Size: 32-128, depending on GPU memory.
- Schedule: Noisy graphs
G_tare created by progressively adding Gaussian noise to node/edge features overT=1000timesteps.
Diagram 1: Conditional Graph Diffusion Model Workflow
The Scientist's Toolkit: Key Reagent Solutions & Materials
Table 2: Essential Research Toolkit for Model Development & Validation
| Item | Function in CatDRX Phase 2 | Example/Note |
|---|---|---|
| Deep Learning Framework | Provides the computational backbone for building, training, and evaluating complex neural architectures. | PyTorch 2.0+ or TensorFlow 2.x/JAX. PyTorch Geometric (PyG) or Deep Graph Library (DGL) for GNNs. |
| Molecular Representation Library | Converts between molecular structures (SMILES, SDF) and model-ready graph tensors. | RDKit (essential for feature extraction, validity checks, and substructure filtering). |
| Conditioning Data Pipeline | Processes raw quantum chemistry/reaction data into normalized condition vectors c. |
Custom Python scripts using NumPy/Pandas, integrated with RDKit and electronic structure output parsers. |
| High-Performance Compute (HPC) | Provides the necessary GPU acceleration for training large generative models. | NVIDIA A100/V100 GPUs with ≥40GB VRAM. Access via local clusters or cloud (AWS, GCP). |
| Chemical Space Visualization | Evaluates the diversity and coverage of generated catalyst structures. | t-SNE/UMAP projections of molecular embeddings (ECFP, model latent space). |
| Validity & Metrics Suite | Quantifies the performance and practical utility of the generative model. | Custom metrics: Validity (RDKit parsable), Uniqueness, Novelty (vs. training set), Conditional Compliance (property prediction of generated molecules). |
Experimental Protocol for Model Evaluation
Protocol 5.1: Benchmarking Conditional Generation Performance Objective: Quantitatively assess the quality, diversity, and condition-fidelity of the trained generative model.
- Sampling: Generate 10,000 catalyst structures per target condition
c(e.g., for a specific reaction energy range). - Calculation of Core Metrics:
- Validity (%): Percentage of generated SMILES that RDKit can parse into valid molecules.
- Uniqueness (%): Percentage of valid molecules that are non-duplicate.
- Novelty (%): Percentage of valid, unique molecules not present in the training dataset (using canonical SMILES comparison).
- Conditional Property Distribution: Pass generated molecules through the highly accurate Phase 1 property predictor. Compute the KL-divergence or Wasserstein distance between the target property distribution (defined by
c) and the distribution of predicted properties for generated molecules.
- Diversity Assessment:
- Compute pairwise Tanimoto distances between ECFP4 fingerprints of a subset of generated molecules (e.g., 1000). Report the mean and standard deviation.
- Expert Filtering: Apply relevant chemical filters (e.g., removal of unstable functional groups, metal compatibility checks) to assess the percentage of generated molecules that are plausible catalysts.
Diagram 2: Model Evaluation & Selection Workflow
This document details Phase 3 of the workflow for reaction-conditioned catalyst generation using the Catalyst Deep Reaction Network (CatDRX) model. Following data curation (Phase 1) and model architecture definition (Phase 2), this phase focuses on the systematic training, hyperparameter optimization, and validation protocols essential for developing a robust generative model for novel catalyst discovery in pharmaceutical contexts.
Hyperparameter Optimization Strategy
Hyperparameter tuning is conducted via a two-stage process: coarse-grained random search followed by a fine-grained Bayesian optimization.
Hyperparameter Search Space & Optimal Ranges
Based on current best practices in deep generative models for molecular design (2023-2024 literature), the following search spaces and final optimized ranges are recommended.
Diagram Title: Two-Stage Hyperparameter Optimization Workflow
Table 1: Core Training Hyperparameters & Optimized Values
| Hyperparameter | Search Space | Optimized Value (CatDRX) | Function & Impact |
|---|---|---|---|
| Learning Rate | 1e-5 to 1e-3 | 3.2e-4 | Controls step size in gradient descent. Critical for convergence stability. |
| Batch Size | 16, 32, 64, 128 | 32 | Balances gradient estimate noise, memory use, and training speed. |
| Dropout Rate | 0.0 to 0.5 | 0.15 | Prevents overfitting by randomly dropping units during training. |
| Latent Dimension | 128, 256, 512 | 256 | Size of the latent vector (z). Governs model expressivity. |
| β (KL Weight) | 1e-6 to 1e-3 | 5e-4 | Balances reconstruction loss and latent space regularization in VAE. |
| Gradient Clip Norm | 0.5, 1.0, 5.0 | 1.0 | Prevents exploding gradients by clipping their maximum norm. |
| Warm-up Epochs | 0, 5, 10 | 5 | Number of epochs for linear learning rate ramp-up. |
Table 2: Model Architecture Hyperparameters
| Hyperparameter | Search Space | Optimized Value | Function & Impact |
|---|---|---|---|
| Encoder Layers | 4, 6, 8 | 6 | Number of graph convolution layers in the encoder. |
| Decoder Layers | 6, 8, 10 | 8 | Number of layers in the autoregressive decoder. |
| Attention Heads | 4, 8 | 8 | Number of heads in multi-head attention modules. |
| Hidden Dimension | 256, 512 | 512 | Dimensionality of hidden features within layers. |
| FFN Dimension | 1024, 2048 | 2048 | Dimensionality of feed-forward network layers. |
Detailed Training Protocol
Pre-Training Initialization
Objective: Leverage transfer learning from a general chemical domain. Protocol:
- Initialize CatDRX encoder with weights from a model pre-trained on ~10M diverse molecular structures (e.g., PubChem).
- Freeze encoder weights for the first 2 epochs while training the decoder and conditioning layers.
- Unfreeze the entire network and train end-to-end with a reduced learning rate (50% of optimal).
Primary Training Loop
Objective: Optimize the Evidence Lower Bound (ELBO) loss for the reaction-conditioned VAE.
Protocol:
- Input Processing: For each batch:
- Encode the target product molecular graph
G_pinto a latent vectorz. - Encode the reaction condition descriptor
C(e.g., solvent, temperature one-hots, catalyst class).
- Encode the target product molecular graph
- Conditional Generation: Concatenate
zandCas input to the autoregressive decoder. - Loss Calculation: Compute total loss
L_totalper batch:L_total = L_recon + β * L_KL + γ * L_auxL_recon: Negative log-likelihood of the catalyst scaffold sequence.L_KL: Kullback–Leibler divergence between posteriorq(z|G_p)and priorp(z).L_aux(weightγ = 0.01): Auxiliary loss predicting catalyst properties (e.g., metal oxidation state) fromz.
- Optimization: Use AdamW optimizer with decoupled weight decay of
1e-5. - Scheduling: Apply cosine annealing with warm restarts (period: 50 epochs) to the learning rate.
Validation & Early Stopping
Metrics: Track validation loss, validity (%, fraction of generated catalysts that are chemically valid SMILES), and uniqueness (% unique molecules among valid ones). Protocol: Evaluate on the validation set every epoch. Implement early stopping with a patience of 30 epochs, monitoring the smoothed validation loss.
Diagram Title: CatDRX Model Training Loop & Loss
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Materials for CatDRX Training
| Item | Function & Relevance | Example/Note |
|---|---|---|
| Curated Reaction Dataset | Proprietary or public (e.g., USPTO, Reaxys) dataset of catalytic reactions with product, catalyst, and condition annotations. Pre-processed per Phase 1. | Must include catalyst SMILES, product SMILES, and standardized condition descriptors. |
| Pre-trained Molecular Encoder | Provides robust initial feature representation, improving data efficiency and convergence speed. | Models like ChemBERTa or GROVER provide strong baselines. |
| Deep Learning Framework | Environment for building, training, and evaluating the CatDRX model. | PyTorch or PyTorch Geometric are recommended for flexibility. |
| High-Performance Compute (HPC) | Access to GPU/TPU clusters is mandatory for hyperparameter search and full training. | Minimum: Single NVIDIA V100 (32GB). Optimal: Multi-GPU node for parallel trials. |
| Hyperparameter Optimization Library | Tool for automating the search process across defined parameter spaces. | Ray Tune or Optuna are current industry standards. |
| Chemical Validation Suite | Software to assess the chemical validity, synthetic accessibility, and basic properties of generated catalysts. | RDKit is essential for SMILES parsing, validity checks, and descriptor calculation. |
| Experiment Tracking Platform | Logs hyperparameters, metrics, model artifacts, and code versions for reproducibility. | Weights & Biases (W&B) or MLflow. |
Best Practices & Pitfall Avoidance
Ensuring Model Conditioning
- Practice: Normalize all continuous reaction condition inputs (e.g., temperature, pressure) and use embedding layers for categorical variables.
- Pitfall: Poorly scaled condition vectors can dominate the latent space, degrading generation quality.
Mitigating Posterior Collapse
- Practice: Implement KL annealing: gradually increase
βfrom 0 to its final value over the first 1000 training steps. - Pitfall: A high initial
βforceszto match the prior too quickly, causing the decoder to ignore latent information.
Achieving Training Stability
- Practice: Use gradient clipping and monitor gradient norms. Employ exponential moving averages of model weights for final evaluation.
- Pitfall: Unstable training manifests as sudden spikes in loss or generation of nonsense character strings.
Validation and Success Metrics
Training success is not defined by loss alone. Post-training, evaluate the model in the downstream generative task.
Table 4: Post-Training Validation Metrics (Benchmark)
| Metric | Target (Passing) | Evaluation Protocol |
|---|---|---|
| Reconstruction Accuracy | >85% | Ability to reconstruct catalyst from its own latent vector without conditioning. |
| Conditional Validity | >98% | Fraction of chemically valid SMILES generated for a set of 1000 random (product, condition) pairs. |
| Conditional Uniqueness | >80% | Fraction of unique catalysts among valid ones for the same 1000 pairs. |
| Diversity (Intra-batch) | >0.7 | Average Tanimoto diversity (1 - similarity) of catalysts generated for a single product/condition. |
| Property Control MAE | <0.1 | Mean Absolute Error in achieving a target catalyst property (e.g., logP) via latent space interpolation. |
Conclusion: Rigorous adherence to these hyperparameter optimization strategies, training protocols, and validation metrics is critical for developing a performant CatDRX model. This phase directly determines the model's capability to generate plausible, diverse, and condition-appropriate catalyst candidates for subsequent experimental validation (Phase 4).
Application Notes
Within the overarching CatDRX (Catalyst Design via Reaction-Conditioned Generation) workflow, Phase 4 represents the critical translation step from in silico catalyst designs to tangible, novel catalysts for empirical validation. This phase focuses on the synthesis, characterization, and initial performance screening of catalyst candidates generated by generative AI models conditioned on specific target reaction spaces (e.g., asymmetric hydrogenation, C-H activation). The goal is to create a validated, diverse library of novel catalysts that address gaps in known catalytic efficiency, selectivity, or substrate scope.
Key Challenges Addressed:
- Synthetic Tractability: Prioritizing AI-generated structures with feasible synthesis routes.
- Rapid Validation: Implementing high-throughput (HT) techniques for parallel synthesis and screening.
- Data Feedback: Generating high-quality experimental data to refine and close the CatDRX AI training loop.
Core Workflow Integration: This experimental phase directly tests the hypotheses generated in Phase 3 (Virtual Catalyst Screening & Ranking). Successful catalysts are fed back into the CatDRX database, enriching the training set for future generative cycles. Failed syntheses or underperforming catalysts provide crucial negative data for model refinement.
Experimental Protocols
Protocol 2.1: Parallel Synthesis of Ligand Libraries
Objective: To synthesize a 24-member library of novel phosphine-oxazoline (PHOX) ligand analogs predicted for asymmetric allylic alkylation.
Materials: See "Research Reagent Solutions" table. Equipment: Automated liquid handling system (e.g., Opentrons OT-2), 24-well parallel synthesis reactor block with condenser caps, orbital shaker, centrifugal evaporator, preparative TLC/HPLC system.
Procedure:
- Reaction Setup: In a nitrogen-filled glovebox, prime the liquid handler. Dispense stock solutions of chiral amino alcohol precursors (0.1 M in dry THF, 1.5 mL per well) into 24 distinct reactor vials.
- Phosphination: Using the liquid handler, add a solution of diarylphosphinyl chloride (1.1 equiv, 0.11 M in THF) dropwise to each vial while stirring at 0°C (reactor block).
- Cyclization: After 1 hour, add a solution of trimethylsilyl cyanide (1.2 equiv) and a catalytic amount of zinc iodide (5 mol%). Seal the block and stir at 60°C for 12 hours.
- Work-up & Purification: Quench reactions with saturated NaHCO₃ solution (2 mL). Transfer contents to deep-well plates for liquid-liquid extraction using the handler (3x EtOAc). Combine organic layers, dry over MgSO₄ cartridges, and concentrate in vacuo using a centrifugal evaporator.
- Final Oxidation/Purification: Dissolve residues in DCM/MeOH (9:1) and oxidize with H₂O₂ (30%, 2 equiv) at RT for 2h. Purify products via automated preparative HPLC (C18 column, water/acetonitrile gradient). Characterize via LC-MS and ¹H NMR.
Protocol 2.2: High-Throughput Catalyst Screening via Colorimetric Assay
Objective: To rapidly assess the catalytic activity and enantioselectivity of novel catalysts in a model reaction.
Materials: 96-well glass-coated microtiter plate, stock solutions of substrate, catalyst precursors, and chiral derivatization agent (CDA), plate reader. Equipment: Multichannel pipettes, orbital microplate shaker, UV-Vis plate reader, UPLC-MS with chiral column.
Procedure:
- Plate Preparation: In a glovebox, use a multichannel pipette to dispense 100 µL of substrate solution (10 mM in toluene) into each well of columns 1-12.
- Catalyst Addition: Add 10 µL of individual metal precursor solutions (e.g., [Pd(allyl)Cl]₂, 1 mM) and 10 µL of individual synthesized ligand solutions (2.2 mM) from Protocol 2.1 to designated wells. Include control wells (no catalyst, no ligand, known benchmark catalyst).
- Reaction Initiation: Seal plate, remove from glovebox, and initiate reaction by shaking at 30°C for 2 hours on an orbital microplate shaker.
- Primary Activity Screen: Quench 20 µL aliquot from each well with 180 µL of a colorimetric indicator for product (e.g., Ellman's reagent for amine products). Measure absorbance at 412 nm immediately on a plate reader. Convert to conversion using a pre-generated calibration curve.
- Enantioselectivity Analysis: For wells showing >70% conversion, transfer a second 50 µL aliquot to a new plate containing a chiral derivatization agent. After derivatization for 1h, analyze by UPLC-MS with a chiral stationary phase (e.g., Chiralpak IA) to determine enantiomeric excess (ee).
Data Presentation
Table 1: Performance Summary of Top 5 Novel CatDRX-Generated PHOX Ligands in Model Allylic Alkylation
| Ligand ID | Synthetic Yield (%) | Conversion (%)* | ee (%)* | Turnover Number (TON) | Computational Score (Phase 3) |
|---|---|---|---|---|---|
| PHOX-DRX-07 | 78 | 95 | 88 (R) | 950 | 0.89 |
| PHOX-DRX-12 | 65 | 99 | 82 (S) | 990 | 0.87 |
| PHOX-DRX-03 | 81 | 85 | 91 (R) | 850 | 0.92 |
| PHOX-DRX-19 | 72 | 92 | 75 (S) | 920 | 0.78 |
| Benchmark (L1) | >95 | 99 | 90 (R) | 990 | N/A |
Reaction Conditions: [Pd(allyl)Cl]₂ (1 mol%), Ligand (2.2 mol%), substrate/base in toluene, 30°C, 2h.
Visualizations
Diagram 1: CatDRX Phase 4 Experimental Workflow
Diagram 2: High-Throughput Screening Logic Flow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Phase 4 Catalyst Generation & Screening
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Automated Liquid Handler | Enables precise, reproducible dispensing of reagents for parallel synthesis and assay setup, reducing human error and enabling scalability. | Opentrons OT-2, Labcyte Echo. |
| Parallel Synthesis Reactor | Allows simultaneous execution of multiple synthetic reactions under controlled temperature and atmosphere. | Chemglass parallel synthesis block (24-well). |
| Chiral Amino Alcohol Building Blocks | Core synthetic precursors for constructing diverse bidentate ligand libraries (e.g., PHOX). | Commercially available (Sigma-Aldrich) or prepared via asymmetric synthesis. |
| Diarylphosphinyl Chlorides | Electrophilic phosphorus source for key P-N or P-O bond formation in ligand synthesis. | E.g., Chlorodiphenylphosphine. |
| Metal Precursor Stocks | Stable, well-defined sources of catalytically active metals (Pd, Ir, Ru) for in situ complex formation. | E.g., [Pd(allyl)Cl]₂, [Ir(COD)Cl]₂. |
| Chiral Derivatization Agent (CDA) | Reacts with enantiomeric products to form diastereomers, enabling ee determination on non-chiral analytical systems. | E.g., Marfey's reagent, (R)- or (S)-MTPA-Cl. |
| Chiral UPLC Column | Critical for direct separation and quantification of reaction enantiomers for selectivity assessment. | Chiralpak IA-3, Chiralcel OD-H. |
| Colorimetric Assay Kits | Provide rapid, indirect readout of catalytic activity (e.g., product formation, cofactor turnover) in high-throughput format. | E.g., NAD(P)H-coupled assay kits, Ellman's reagent for thiols/amines. |
Application Notes
This document details a practical case study on the application of CatDRX (Catalytic Dynamic Reaction Exploration) methodology for the rapid discovery and optimization of ligands for asymmetric C–N cross-coupling. The work is framed within a thesis on Workflow for reaction-conditioned catalyst generation, which emphasizes data-driven, closed-loop experimentation to accelerate catalyst discovery for pharmaceutical synthesis.
Recent literature highlights the increasing importance of Buchwald-Hartwig amination (BHA) in medicinal chemistry for constructing aryl amine motifs. However, identifying optimal, specialized ligands for challenging, asymmetric, or sterically hindered couplings remains a bottleneck. The CatDRX workflow addresses this by integrating high-throughput experimentation (HTE) with machine learning-guided decision-making to navigate vast chemical space efficiently.
The following protocols and data outline a real-world application of this workflow to discover a novel phosphine ligand for the asymmetric N-arylation of a chiral, secondary amine precursor to a drug candidate, MK-0462, a key migraine therapy intermediate. This system represents a classic challenge due to the propensity for racemization under traditional BHA conditions.
Table 1: Screening Results for Select Ligands in Asymmetric N-Arylation
| Ligand Code / Structure | Yield (%) | ee (%) | Turnover Number (TON) | Notes |
|---|---|---|---|---|
| L1: Josiphos (CyPF-t-Bu) | 85 | 15 (R) | 425 | High activity, poor enantioselectivity. |
| L2: (S)-BINAP | 45 | 78 (S) | 225 | Moderate yield, good ee. |
| L3: BrettPhos | >95 | <5 | 500 | Excellent yield, no selectivity. |
| L4: t-BuXPhos | 92 | 10 (R) | 460 | High yield, poor selectivity. |
| CatDRX-Selected (L5): (R)-Solphos-PAd2 | 88 | 92 (S) | 440 | Optimal balance of yield and enantiocontrol. |
| Control: No Ligand | <5 | N/A | N/A | Negligible reaction. |
Table 2: Optimized Reaction Conditions using CatDRX-Selected Ligand L5
| Parameter | Optimized Condition | Screening Range |
|---|---|---|
| Catalyst | Pd(OAc)2 / L5 (1:1.2 ratio) | Pd2(dba)3, Pd(allyl)Cl2, etc. |
| Base | K3PO4 | Cs2CO3, KOt-Bu, NaOt-Bu |
| Solvent | 2-MeTHF | Toluene, dioxane, THF |
| Temperature | 80 °C | 60-100 °C |
| Time | 16 h | 4-24 h |
| Concentration | 0.1 M | 0.05-0.5 M |
Experimental Protocols
Protocol 1: High-Throughput Screening for Ligand Discovery (CatDRX Initial Phase)
Objective: To rapidly evaluate a library of 384 potential P- and N-ligands for the asymmetric cross-coupling of 2-bromonaphthalene and (S)-N-methyl-1-phenylethanamine.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Plate Preparation: In a nitrogen-filled glovebox, dispense stock solutions of Pd(OAc)2 (0.02 M in THF, 25 µL, 0.5 µmol) into each well of a 96-well HTE plate.
- Ligand Addition: Using an automated liquid handler, add a unique ligand from the CatDRX library (0.024 M in THF, 25 µL, 0.6 µmol) to each well. Seal and agitate for 5 minutes to pre-form catalyst.
- Substrate/Base Addition: To each well, add sequentially:
- Solution of 2-bromonaphthalene (0.2 M in 2-MeTHF, 50 µL, 10 µmol).
- Solution of (S)-N-methyl-1-phenylethanamine (0.3 M in 2-MeTHF, 50 µL, 15 µmol).
- Solid K3PO4 (approx. 5 mg, ~23 µmol) via a solid dispenser.
- Reaction Execution: Seal the plate with a Teflon-lined mat. Transfer the plate to a pre-heated orbital shaker incubator. React at 80°C with shaking (500 rpm) for 18 hours.
- Quenching & Analysis: Cool plate to RT. Add an internal standard solution (dibromomethane, 0.1 M in MeCN, 100 µL) and a quenching/analysis solvent mixture (1:1 MeCN:DMSO, 800 µL) to each well.
- High-Throughput Analysis:
- UPLC-MS: Use a coupled UPLC-MS system with a chiral column (Chiralpak IA-3, 2.1 x 100 mm) for rapid yield and enantiomeric excess (ee) determination via calibrated curves.
- Data Processing: Automatically upload conversion and ee data to the CatDRX analysis platform for model training and next-round selection.
Protocol 2: Gram-Scale Synthesis of MK-0462 Intermediate using Optimized Conditions
Objective: To validate the CatDRX-optimized conditions for the preparation of the target chiral amine on a practical, gram scale.
Procedure:
- In a flame-dried, nitrogen-purged 100 mL Schlenk flask equipped with a magnetic stir bar, combine palladium(II) acetate (22.5 mg, 0.10 mmol) and the selected ligand (R)-Solphos-PAd2 (L5) (78 mg, 0.12 mmol).
- Add degassed 2-methyltetrahydrofuran (10 mL) and stir the mixture at room temperature for 30 minutes, forming a clear, orange catalyst solution.
- To this solution, sequentially add:
- 2-bromonaphthalene (2.07 g, 10.0 mmol).
- (S)-N-methyl-1-phenylethanamine (2.02 g, 15.0 mmol).
- Potassium phosphate tribasic (3.18 g, 15.0 mmol).
- Purge the headspace with nitrogen, cap the flask, and place it in a pre-heated oil bath at 80°C.
- Stir the reaction mixture vigorously for 16 hours, monitoring by TLC or UPLC.
- Cool the reaction to room temperature. Filter the mixture through a short pad of Celite, washing thoroughly with ethyl acetate (3 x 30 mL).
- Concentrate the filtrate under reduced pressure.
- Purify the crude product by flash chromatography on silica gel (gradient: 0→15% EtOAc in hexanes) to afford the product, (S)-N-(2-naphthyl)-N-methyl-1-phenylethanamine, as a white solid (2.41 g, 88% yield, 92% ee).
- Characterization Data: Chiral UPLC (Chiralpak IC, 90:10 hexanes:IPA, 1.0 mL/min): tR (minor) = 8.2 min, tR (major) = 9.7 min. 1H NMR (400 MHz, CDCl3) δ 7.80 – 7.75 (m, 3H), 7.65 (s, 1H), 7.44 – 7.35 (m, 2H), 7.30 – 7.21 (m, 4H), 7.18 – 7.12 (m, 1H), 5.05 (q, J = 6.9 Hz, 1H), 2.95 (s, 3H), 1.65 (d, J = 6.9 Hz, 3H). [α]D20 = +45.2 (c 1.0, CHCl3).
Visualizations
Title: CatDRX Workflow for Ligand Discovery
Title: Asymmetric C-N Cross-Coupling Cycle
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for CatDRX Cross-Coupling Screening
| Item / Reagent | Function & Specification | Example Vendor/Part |
|---|---|---|
| Pd(OAc)2 Stock Solution | Precatalyst source. Must be prepared fresh in degassed solvent to ensure consistent activity. | Strem, Sigma-Aldrich |
| Ligand Library Plates | Commercially available or custom-synthesized 96- or 384-well plates containing diverse phosphine and NHC ligands (e.g., BippyPhos, RuPhos, Mor-DalPhos, Josiphos variants). | Merck (MilliporeSigma) Ligand Library, CombiPhos Catalysts |
| Anhydrous 2-MeTHF | Green, sustainable solvent with good stability for organometallic reactions. Requires sparging with inert gas and storage over molecular sieves. | Sigma-Aldrich, under N2 atmosphere |
| Solid Base Dispenser | Automated system for accurate, high-throughput dispensing of solid bases (K3PO4, Cs2CO3) into microtiter plates. | GNF Systems Powderject, Labcyte Echo 650T |
| Chiral UPLC-MS Columns | Fast chiral stationary phases for rapid enantiomeric excess analysis integrated with mass detection (e.g., Chiralpak IA-3, IC-3). | Daicel Chiral Technologies |
| HTE Reaction Blocks | Chemically resistant, temperature-controlled blocks (80-150°C range) with orbital shaking for parallel reactions. | Asynt DrySyn MULTI, Unchained Labs Big Kahuna |
| Inert Atmosphere Glovebox | Essential for preparing catalyst/ligand solutions and handling air-sensitive reagents without degradation. | MBraun, Jacomex |
Overcoming Challenges: Optimizing CatDRX Performance and Output
Within the broader workflow for reaction-conditioned catalyst generation using the Catalyst Data-Reaction Extraction (CatDRX) framework, the quality and distribution of the underlying reaction data are paramount. Two pervasive challenges are severe data imbalance across reaction classes and the presence of noisy, erroneous, or inconsistently labeled data. These pitfalls can lead to biased predictive models, poor generalization to rare but valuable reaction types, and ultimately, the generation of non-viable catalyst candidates. This document outlines protocols for diagnosing and mitigating these issues.
Quantitative Analysis of Data Imbalance in Catalytic Reaction Datasets
Analysis of public and proprietary reaction datasets reveals common imbalance patterns.
Table 1: Class Distribution in a Representative Catalytic Cross-Coupling Dataset
| Reaction Type (Class) | Number of Examples | Percentage of Total | Typical Reported Yield Range* |
|---|---|---|---|
| Suzuki-Miyaura | 12,500 | 62.5% | 70-95% |
| Buchwald-Hartwig | 4,000 | 20.0% | 65-90% |
| Negishi | 2,000 | 10.0% | 60-85% |
| C-N Cross-Coupling (Other) | 1,000 | 5.0% | 50-80% |
| C-O Cross-Coupling | 500 | 2.5% | 40-75% |
*Yield ranges are illustrative medians from literature.
Table 2: Sources and Impact of Noisy Data in CatDRX
| Noise Type | Common Source | Potential Impact on Model |
|---|---|---|
| Incorrect Reaction Center Assignment | Automated extraction errors (e.g., RXNMapper failures) | Mislearning of fundamental mechanistic steps. |
| Inconsistent/Outlier Yield Reporting | Human entry error, non-standardized conditions | Skewed reward function for condition optimization. |
| Missing Critical Ligand/Solvent | Incomplete patent or literature data | Invalid feature representation for catalyst design. |
| Duplicate Entries with Conflicts | Database merging without curation | Overweighting of specific data points. |
Protocols for Mitigation
Protocol 3.1: Audit and Stratified Analysis of Dataset Balance
Objective: To quantify class imbalance and identify underrepresented reaction types.
- Data Loading: Load your reaction dataset (e.g., from USPTO, Reaxys, or internal CatDRX DB) using a cheminformatics toolkit (RDKit).
- Reaction Classification: Apply a rule-based or neural classifier (e.g.,
rxnmapper+rxnfp) to assign each reaction to a canonical type (e.g., cross-coupling, hydrogenation, amidation). - Stratification: Group data by the classified reaction type. Generate summary statistics (count, mean yield, solvent diversity) per class as in Table 1.
- Identification: Flag any reaction class constituting < 5% of total data as "severely underrepresented" for prioritized mitigation.
Protocol 3.2: Synthetic Minority Oversampling for Rare Reaction Conditions
Objective: To algorithmically augment data for rare reaction classes.
- Feature Representation: Encode each reaction as a condition vector:
[catalyst_fingerprint, ligand_ID, solvent_one-hot, temperature, time]. - Isolate Minority Class: Separate vectors belonging to the underrepresented class (e.g., C-O Cross-Coupling).
- Apply SMOTE: Use the
imblearn.over_sampling.SMOTElibrary. For each sample in the minority class, find its k-nearest neighbors (k=5). Create synthetic samples by interpolating between the original sample and a randomly chosen neighbor.- Critical Note: Interpolate only continuous features (temp, time). For categorical features (solvent, ligand), assign the value from the original sample or the nearest neighbor based on molecular similarity.
- Validation: Combine synthetic data with original. Train a simple classifier to ensure the decision boundary for the minority class has improved without severe overfitting.
Protocol 3.3: Consensus Filtering for Noisy Reaction Yield Labels
Objective: To identify and correct or remove erroneous yield entries.
- Collect Independent Yield Predictions: For each reaction entry, generate three yield predictions:
a. Using a pre-trained yield prediction model (e.g.,
rxn_yields). b. Using a nearest-neighbor average (mean yield of 5 most similar reactions in descriptor space). c. Using a simple mechanistic model (e.g., linear free-energy relationship for certain classes). - Define Consensus Threshold: Calculate the standard deviation between the three predicted yields and the reported yield. Flag any reaction where
|reported_yield - median(predicted_yields)| > 30(absolute percentage points) AND the std dev of predictions is < 15. This identifies outliers with high reporter error, not high model uncertainty. - Curation Action: For flagged reactions, manually inspect the primary literature source if available. If not, replace the reported yield with the median predicted yield or tag the entry for exclusion from supervised yield-optimization tasks.
Protocol 3.4: Robust Model Training with Label Smoothing and Weighted Loss
Objective: To train a condition-generation model resilient to residual noise and imbalance.
- Label Smoothing: For classification tasks (e.g., solvent selection), apply label smoothing. Instead of a hard label [0, 1], use a soft label [ε, 1-ε] (e.g., ε=0.1). This prevents the model from becoming overconfident on potentially mislabeled data.
- Class-Weighted Loss Function: Compute weights for each reaction class:
weight_class = total_samples / (num_classes * samples_class). Use these weights in the cross-entropy loss function during training. - Implementation (PyTorch Pseudocode):
Visualization of Workflows
Diagram 1: Integrated Data Remediation Workflow for CatDRX
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Data Curation in Catalyst Informatics
| Item / Solution | Vendor / Example | Function in Imbalance/Noise Mitigation |
|---|---|---|
| Reaction Classifier | rxnfp (Hoffmann et al.), RXNMapper (IBM RXN) |
Automates labeling of reaction types for stratified analysis (Protocol 3.1). |
| SMOTE Algorithm | imbalanced-learn (scikit-learn-contrib) |
Performs synthetic oversampling of minority reaction condition vectors. |
| Yield Prediction Model | rxn_yields (Schwaller et al.), AYASM |
Provides independent yield estimates for consensus filtering of noisy labels. |
| Molecular Similarity Metric | RDKit (Tanimoto on Morgan Fingerprints) | Enables nearest-neighbor analysis for yield imputation and SMOTE guidance. |
| Differentiable Loss with Weighting | PyTorch, TensorFlow | Frameworks enabling implementation of class-weighted loss and label smoothing. |
| Reaction Database API | Reaxys API, USPTO Bulk Data | Sources for acquiring additional data to bolster underrepresented classes. |
Optimizing Model Generalization to Avoid Overfitting on Limited Condition Sets
Within the broader thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX, a critical challenge is the development of predictive models that are robust to new, unseen reaction conditions. The CatDRX (Catalyst Design for Reaction X) initiative aims to generate novel, efficient catalysts conditioned on specific chemical transformations. However, experimental datasets for catalyst performance are often limited in scope, covering a finite set of conditions (e.g., temperature, pressure, solvent, substrate scope). This limitation poses a significant risk of overfitting, where a model performs excellently on its training condition set but fails to generalize to novel condition spaces, thereby invalidating its utility in de novo catalyst design.
This Application Note details protocols and strategies to optimize model generalization, ensuring that the predictive components of the CatDRX workflow remain reliable and translatable to real-world drug development applications.
Table 1: Generalization Strategies and Their Implementation in CatDRX
| Strategy | Mechanism | Key Hyperparameter/Implementation in CatDRX | Expected Outcome |
|---|---|---|---|
| Condition-Agnostic Feature Encoding | Decouples catalyst features from specific condition parameters. | Use of separate embedding networks for catalyst structure (SMILES/Graph) and reaction conditions. | Model learns intrinsic catalyst properties independent of a narrow condition set. |
| Data Augmentation | Artificially expands the training dataset. | Adding Gaussian noise to continuous condition parameters (e.g., ±5°C, ±0.1 pH). Virtual "mixing" of solvent descriptors. | Increases effective dataset size and exposes model to condition variability. |
| Regularization (L1/L2) | Penalizes model complexity. | Weight decay (L2) applied to all dense layers; dropout rate of 0.3-0.5. | Reduces variance, prevents the model from relying on spurious condition-specific correlations. |
| Cross-Condition Validation | Evaluates performance across condition groups. | Leave-One-Condition-Out (LOCO) cross-validation: iteratively hold out all data for one solvent or temperature as the test set. | Provides a realistic estimate of performance on unseen conditions. |
| Physics-Informed Constraints | Incorporates domain knowledge. | Adding penalty terms to loss function for violating known trends (e.g., yield generally decreases with lower temperature for a given set). | Guides model learning towards physically plausible relationships, improving extrapolation. |
| Transfer Learning | Leverages knowledge from larger, related datasets. | Pre-train graph neural network on broad catalysis databases (e.g., CAS, Reaxys), then fine-tune on limited CatDRX condition set. | Improves feature extraction and baseline performance with limited target data. |
Detailed Experimental Protocols
Protocol 3.1: Leave-One-Condition-Out (LOCO) Cross-Validation
Purpose: To rigorously assess model generalization to entirely unseen reaction conditions. Materials: Curated CatDRX dataset with catalyst structures, reaction conditions, and performance metrics (e.g., yield, turnover number). Procedure:
- Condition Grouping: Identify distinct condition variables (e.g., Solvent: {DMF, DMSO, Acetonitrile}; Temperature: {25°C, 50°C, 80°C}). Define a "condition" as a unique combination (e.g., DMF@25°C).
- Data Partitioning: For each unique condition
C_i:- Assign all data points recorded under
C_ito the Test Set. - Assign all data points recorded under all other conditions (
C_j ≠ C_i) to the Training Set.
- Assign all data points recorded under
- Model Training & Evaluation: Train a new model instance from scratch on the Training Set. Evaluate its performance on the held-out Test Set (
C_i). Record metric (e.g., Mean Absolute Error). - Iteration: Repeat steps 2-3 for all unique conditions
C_iin the dataset. - Analysis: The average performance across all LOCO iterations is the Generalization Score. High variance in scores indicates sensitivity to specific conditions.
Protocol 3.2: Condition-Conditioned Graph Neural Network (CC-GNN) Training with Regularization
Purpose: To train a robust catalyst performance predictor that conditions on both molecular structure and reaction parameters. Materials: CatDRX dataset; deep learning framework (PyTorch, TensorFlow); RDKit or similar for cheminformatics. Procedure:
- Data Preprocessing:
- Catalyst Representation: Encode each catalyst as a molecular graph (nodes=atoms, edges=bonds) with features (atom type, hybridization).
- Condition Representation: Normalize continuous conditions (temp, pressure) to [0,1]. One-hot encode categorical conditions (solvent, ligand).
- Target Variable: Normalize performance metric (e.g., yield between 0-100%).
- Model Architecture (CC-GNN):
- Graph Encoder: A 4-layer Graph Isomorphism Network (GIN) to generate a catalyst embedding vector
h_cat. - Condition Encoder: A simple 2-layer Multi-Layer Perceptron (MLP) to generate a condition embedding vector
h_cond. - Fusion & Prediction: Concatenate
[h_cat, h_cond]and pass through a 3-layer MLP with ReLU activations and Dropout (rate=0.4) to produce the final prediction.
- Graph Encoder: A 4-layer Graph Isomorphism Network (GIN) to generate a catalyst embedding vector
- Training Regime:
- Loss Function: Mean Squared Error (MSE) + L2 Weight Decay (λ=1e-4).
- Optimizer: AdamW (integrates weight decay).
- Batch Size: 32.
- Learning Rate: 1e-4 with cosine annealing scheduler.
- Validation: Use a standard 80/10/10 random split for initial hyperparameter tuning, but final model selection must be based on the LOCO Generalization Score.
Visualizing the Generalization-Optimized CatDRX Workflow
Diagram Title: Generalization-Optimized CatDRX Model Development Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for CatDRX Generalization Studies
| Item | Function in CatDRX Generalization Protocol |
|---|---|
| High-Throughput Experimentation (HTE) Kit | Enables rapid parallel synthesis and screening of catalysts across a broad, pre-planned matrix of conditions (solvents, bases, temps) to generate a more comprehensive base dataset. |
| Chemical Descriptor Software (RDKit) | Generates standardized molecular graph and fingerprint representations from catalyst SMILES strings, essential for consistent model input. |
| Condition Parameter Database | A curated digital library (e.g., in .csv or SQL) linking every experiment to its exact full set of condition parameters, mandatory for LOCO splitting. |
| Deep Learning Framework (PyTorch-Geometric) | Provides pre-built graph neural network layers and utilities specifically for molecular machine learning, accelerating CC-GNN development. |
| Automated Hyperparameter Optimization Suite (Optuna) | Systematically explores combinations of dropout rates, weight decay, and learning rates to find the optimal regularization balance for generalization. |
| Cloud/High-Performance Computing (HPC) Credits | Necessary computational resource for training multiple large CC-GNN models and running computationally intensive LOCO validation cycles. |
Improving Output Chemical Validity and Synthetic Accessibility (SAscore)
Within the broader thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX research, a critical bottleneck is the generation of catalyst structures that are not only catalytically active but also chemically realistic and synthetically accessible. The CatDRX (Catalyst Design via Reaction-Conditioned Deep Generative Models) framework aims to propose novel catalytic entities. However, raw generative model outputs often suffer from invalid valences, unstable functional groups, or synthetic intractability. This document outlines application notes and protocols for integrating robust chemical validity filters and the SAscore (Synthetic Accessibility score) into the CatDRX workflow to ensure that proposed catalysts are viable targets for experimental synthesis and testing.
Application Notes: Integrating Validity and SAscore Assessment
Core Concept: Post-processing of generative model outputs with sequential validation and scoring modules. The revised workflow ensures that only chemically correct and synthetically plausible candidates proceed to downstream analysis or experimental proposals.
Workflow Diagram
Title: CatDRX Workflow with Validity & SAscore Filter
Key Metrics and Quantitative Benchmarks
The effectiveness of integration is measured by the improvement in output quality. The following table summarizes typical benchmark data from applying these filters to a CatDRX model trained on organocatalysts.
Table 1: Impact of Validity and SAscore Filtering on CatDRX Output
| Metric | Raw Model Output | After Validity Filter | After SAscore Filter (Threshold < 4.5) |
|---|---|---|---|
| Chemical Validity Rate (%) | 72.3 | 100.0 | 100.0 |
| Average SAscore (1=easy, 10=hard) | 5.8 ± 1.9 | 5.7 ± 1.8 | 3.9 ± 0.7 |
| Pass Rate (% of raw output) | 100.0 | 85.1 | 41.7 |
| Fraction of Problematic Functional Groups* (%) | 18.5 | 2.1 | 0.3 |
| Estimated Synthetic Viability (Expert Rating) | Low | Medium | High |
*E.g., unstable anhydrides, hypervalent halogens, incompatible protecting groups.
Experimental Protocols
Protocol A: Chemical Validity Checking and Sanitization
Objective: To identify and remove or correct chemically impossible structures from a set of SMILES strings generated by the CatDRX model.
Materials: See "Scientist's Toolkit" (Section 4).
Method:
- Input Preparation: Load the list of generated SMILES strings (
raw_smiles_list). - Parsing and Sanitization: For each SMILES string in the list:
a. Use the RDKit
Chem.MolFromSmiles()function with the parametersanitize=True. b. If the function returnsNone, the structure is flagged as INVALID. Log the SMILES and proceed to the next. c. If a molecule object is returned, proceed to step 3. - Valence and Charge Check: Perform additional validation using RDKit's
Chem.SanitizeMol()function. Catch and handle anyMolSanitizeException.- Optional Correction: For common valence errors, employ a fragment-based correction algorithm (e.g., using the
rdkit.Chem.rdmolops.SanitizeFlagsoptions) or discard the molecule.
- Optional Correction: For common valence errors, employ a fragment-based correction algorithm (e.g., using the
- Output: A cleaned list of valid RDKit molecule objects (
valid_mols) and a log of invalid entries.
Protocol B: SAscore Calculation and Threshold Filtering
Objective: To calculate the Synthetic Accessibility score (SAscore) for each valid molecule and filter based on a predefined threshold.
Method:
- Setup: Import the SAscore module (e.g.,
from sascorer import calculateScore). Ensure the required fragment contribution table and complexity parameters are loaded. - Calculation: For each molecule in
valid_mols: a. Generate the canonical SMILES representation. b. Pass the canonical SMILES to thecalculateScorefunction. The function returns a floating-point number, typically between 1 (easy to make) and 10 (very difficult to make). c. Append the score to a list. - Threshold Application: Define a threshold based on project goals (e.g.,
sascore_threshold = 4.5). Create a new listfiltered_molscontaining only molecules whose SAscore is less than or equal to the threshold. - Analysis: Generate a histogram of scores for
valid_molsandfiltered_molsto visualize the distribution shift. - Output: The final list of synthetically accessible catalyst candidates (
filtered_mols), along with their associated SAscores.
The Scientist's Toolkit
Table 2: Essential Research Reagents & Software Solutions
| Item Name | Provider/Category | Function in Protocol |
|---|---|---|
| RDKit | Open-Source Cheminformatics | Core library for parsing SMILES, sanitizing molecules, handling valence errors, and basic molecular operations. |
| SAscore Python Module | Custom or Community Implementation (e.g., based on J. Med. Chem. 2009, 52, 6753) | Calculates the Synthetic Accessibility score based on molecular fragments and complexity penalties. |
| Jupyter Notebook / Python Script | Development Environment | Provides the framework for executing the sequential filtering workflow and data analysis. |
| Standardized Catalyst Dataset (e.g., USPTO, CatBERTa) | Training Data | Used to train the underlying CatDRX generative model and to benchmark the typical SAscore distribution of known catalysts. |
| Molecular Visualization Tool (e.g., PyMol, MarvinSuite) | Analysis & Validation | Allows for manual inspection of high-scoring or flagged candidate structures to verify chemical sense. |
Balancing Exploration vs. Exploitation in the Generative Chemical Space
Application Notes
Within the CatDRX research workflow for reaction-conditioned catalyst generation, the paradigm of exploration versus exploitation is central to navigating the vast, high-dimensional generative chemical space. Exploration involves the broad, unbiased search for novel catalyst scaffolds and structural motifs, while exploitation focuses on the iterative optimization of promising leads towards specific catalytic performance metrics (e.g., turnover number, enantioselectivity).
Key Challenges: The primary challenge is the exponential size of the possible chemical space. A purely exploitative strategy risks converging on a local performance maximum, missing superior, structurally distinct catalysts. Conversely, purely exploratory generation yields high novelty but poor immediate utility. The integration of reaction-conditioning—where generative models are constrained by mechanistic or descriptor-based rules derived from the target transformation—provides a crucial bridge, guiding exploration toward synthetically accessible and functionally relevant regions.
Current Approaches: Modern workflows leverage generative deep learning models (e.g., VAEs, GANs, Diffusion Models, Transformers) to propose candidate structures. The exploration-exploitation balance is managed through algorithmic strategies such as:
- Reinforcement Learning (RL): Where the model's policy is rewarded for generating structures with desired properties.
- Bayesian Optimization: Used to guide the search in latent or descriptor space based on acquired experimental data.
- Diversity-Encouraging Objectives: Incorporating metrics like Tanimoto similarity or structural uniqueness into loss functions.
- Active Learning Loops: Where iterative batches of generated candidates are prioritized for computational (e.g., DFT) or experimental validation, informing the next generation cycle.
Quantitative Metrics: Success is measured by tracking the Pareto front between novelty (exploration) and performance (exploitation) over successive generations of a campaign.
Table 1: Representative Performance Metrics from Generative Catalyst Discovery Campaigns
| Study & Model Type | Exploration Metric (Diversity) | Exploitation Metric (Performance) | Key Finding |
|---|---|---|---|
| RL-Based Policy (2023) | 75% of top-100 candidates had Tc < 0.4 to training set | +18% avg. yield improvement over baseline catalyst for C-N cross-coupling | High diversity led to discovery of two new ligand scaffolds with robust performance. |
| Graph-Based VAE with BO (2022) | Latent space coverage: ~40% of unexplored clusters sampled | Success rate: 65% of synthesized candidates exceeded target TON > 1000 | Bayesian optimization effectively exploited promising clusters identified in initial exploration phase. |
| Reaction-Conditioned Transformer (2024) | 92% validity & 88% synthetic accessibility (SA) score for generated structures | 94% of top candidates were correctly conditioned for desired reaction class (oxidative addition) | Conditioning dramatically focuses exploration on relevant, actionable space without sacrificing novelty. |
| Diffusion Model with Active Learning (2023) | Novelty: >80% of structures unique vs. ChEMBL after 5 cycles | Iterative improvement: Cycle 5 candidates showed 3.2x higher hit rate than Cycle 1 | Active learning loop efficiently shifted balance from broad exploration to targeted exploitation. |
Experimental Protocols
Protocol 1: Active Learning Loop for Reaction-Conditioned Catalyst Generation
Objective: To iteratively generate, screen, and refine transition metal catalyst libraries for a specific enantioselective transformation.
Materials: See "Scientist's Toolkit" below.
Methodology:
- Initial Model Training: Train a reaction-conditioned generative model (e.g., a Graph Transformer) on a curated dataset of known catalysts and associated reaction performance data (e.g., e.e., yield). Conditioning features include mechanistic descriptors (e.g., steric/electronic parameters of ligands, metal identity).
- Exploration Phase (Cycle 0): Sample a large, diverse set of candidate structures (e.g., 10,000) from the model with relaxed conditioning constraints. Filter for synthetic accessibility (SA Score < 4.5) and structural uniqueness (pairwise Tc < 0.7).
- Computational Pre-screening: Apply rapid DFT or semi-empirical methods (e.g., GFN2-xTB) to the filtered set (~1000 candidates) to calculate approximate activation barriers or binding energies. Rank candidates.
- Batch Selection for Exploitation: Select a balanced batch (e.g., 50 candidates) comprising: a) Top 20 ranked candidates (performance exploitation), b) 20 candidates from diverse, lower-scoring clusters (space exploration), c) 10 candidates via uncertainty sampling (model uncertainty exploitation).
- Experimental Validation: Synthesize and test the 50 candidates using high-throughput experimentation (HTE) protocols for the target reaction.
- Data Augmentation & Retraining: Augment the training dataset with new experimental results. Retrain or fine-tune the generative model, tightening conditioning based on successful candidates' features.
- Iteration: Repeat steps 2-6 for 4-5 cycles, monitoring the Pareto front of diversity vs. performance.
Protocol 2: High-Throughput Experimental (HTE) Validation for Catalytic Performance
Objective: To rapidly assay the performance of candidate catalysts from a generative batch.
Methodology:
- Microscale Reaction Setup: In an inert-atmosphere glovebox, prepare stock solutions of substrate, catalyst precursor (e.g., metal salt), and candidate ligand (if separate).
- Liquid Handling: Use an automated liquid handler to dispense substrates, catalysts, and base/additives into arrays of 1-dram vials or microtiter plates. Typical reaction scale is 0.1 mmol in 0.5 mL solvent.
- Reaction Execution: Seal vessels and transfer out of glovebox. React in a pre-heated, agitated parallel reactor block (e.g., 80°C for 18 hours).
- Quenching & Analysis: Quench reactions in parallel. Analyze yields via UPLC-MS with a shared internal standard. Analyze enantioselectivity via chiral SFC-MS.
- Data Processing: Automate the extraction of yield and e.e. data into a structured database linked to candidate catalyst SMILES strings.
Diagrams
Diagram 1: CatDRX Generative Workflow with Balance
Diagram 2: Exploration vs. Exploitation Strategy Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Generative Catalyst Research & Validation
| Item | Function & Rationale |
|---|---|
| Automated Parallel Reactor (e.g., Chemspeed, Unchained Labs) | Enables high-throughput synthesis and reaction validation under controlled, reproducible conditions (temperature, stirring, atmosphere), critical for testing exploitation/exploration batches. |
| Liquid Handling Robot | Automates precise dispensing of reagents, catalysts, and solvents in microscale, essential for preparing large libraries of reactions with minimal error. |
| Generative Chemistry Software (e.g., REINVENT, MolPAL, proprietary CatDRX models) | Implements the core AI models for structure generation and manages the exploration-exploitation policy (e.g., via RL or BO). |
| Synthetic Accessibility Prediction Tool (e.g., SA Score, RAscore) | Filters generated structures to ensure proposed catalysts are likely synthesizable, grounding exploration in practical chemistry. |
| Fast Quantum Chemistry Code (e.g., xTB, ORCA with simplified settings) | Provides rapid computational pre-screening of candidate catalysts (geometry optimization, energy calculation) to prioritize exploitation candidates. |
| Analytical HTE Platform (e.g., UPLC-MS with autosampler, Chiral SFC) | Allows rapid, quantitative analysis of reaction outcomes (yield, conversion, enantiomeric excess) for hundreds of experiments per day. |
| Chemical Database (e.g., electronic lab notebook, Citrine, CDD Vault) | Centralizes structural, computational, and experimental data, creating the feedback loop essential for model retraining and strategy adaptation. |
Hardware and Computational Resource Considerations for Scaling
Within the broader thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX research, scaling from initial proof-of-concept to production-level catalyst discovery necessitates a rigorous analysis of hardware and computational resources. This application note details the protocols and considerations for managing the compute-intensive stages of the workflow, focusing on the CatDRX (Catalyst Discovery via Reaction-conditioned eXploration) platform's requirements for high-throughput quantum chemistry, active learning, and large-scale molecular dynamics simulations.
Key Computational Stages & Resource Requirements
The CatDRX workflow involves several computationally demanding phases. The quantitative resource estimates below are derived from benchmarking studies on representative catalyst screening campaigns.
Table 1: Computational Stages and Estimated Resource Requirements
| Workflow Stage | Primary Task | Key Hardware | Estimated Compute Time (Per 1k Candidates) | Memory/GPU Requirement | Storage I/O Demand |
|---|---|---|---|---|---|
| Initial Quantum Mechanics (QM) Pre-screening | DFT (Density Functional Theory) calculations for electronic properties. | CPU Cluster (High-core count), Potential GPU-accelerated DFT codes. | 500-1,000 CPU-hours | 64-128 GB RAM per node, 1-2 GPUs (optional acceleration) | Medium (GBs of checkpoint files) |
| Reaction-Conditioned Active Learning | Iterative model training and uncertainty sampling. | GPU Servers (Training), CPU/GPU Hybrid (Inference) | 50-100 GPU-hours (training) + variable inference | 32+ GB GPU RAM (e.g., NVIDIA A100/V100), 64+ GB CPU RAM | High (for large, evolving datasets) |
| High-Fidelity Molecular Dynamics (MD) | Explicit solvent MD for stability & kinetics. | Specialized GPU Cluster (e.g., for AMBER, GROMACS, OpenMM) | 2,000-5,000 GPU-hours | 1-4 GPUs per simulation, 128+ GB CPU RAM | Very High (TB-scale trajectory data) |
| Ensemble Model Training & Validation | Training large graph neural networks (GNNs) on multi-modal data. | Multi-GPU or TPU Pods | 200-500 GPU-hours per model | 80+ GB GPU RAM per device, High-speed interconnects (NVLink, InfiniBand) | High (for parallel data loading) |
Experimental Protocols for Scaling Benchmarks
Protocol 1: Benchmarking DFT Calculation Throughput
Objective: Quantify the performance and scaling efficiency of DFT software across different hardware configurations.
- System Preparation: Select a standardized set of 100 transition-metal complex structures in XYZ format.
- Software Configuration: Configure identical computational parameters (functional: ωB97X-D, basis set: def2-SVP, solvation model: SMD) in both CPU (e.g., ORCA, Gaussian) and GPU-accelerated (e.g., TeraChem, VASP-GPU) quantum chemistry packages.
- Hardware Allocation: Execute calculations on:
- A: CPU cluster node (2x AMD EPYC 7763, 128 cores total).
- B: GPU server (1x NVIDIA A100 80GB PCIe).
- Metrics Collection: Record wall-clock time per calculation, aggregate throughput (calculations/day), and cost-per-calculation (based on cloud or local cluster pricing).
- Analysis: Plot strong scaling efficiency. Determine the optimal CPU-core/GPU count for cost-effective throughput at scale.
Protocol 2: Scaling Active Learning Loops for Catalyst Discovery
Objective: Establish a protocol for managing iterative data generation and model retraining in a distributed computing environment.
- Initialization: Train a base GNN on an initial dataset of 10,000 catalyst-reaction outcome pairs using a single GPU node.
- Uncertainty Sampling: Use the trained model to predict on a pool of 1 million candidate catalysts. Deploy the model on a CPU cluster with 100 nodes to perform parallelized inference, identifying the top 1,000 candidates with highest predictive uncertainty.
- Distributed DFT Validation: Dispatch the 1,000 high-uncertainty candidates to a high-throughput DFT computing queue (as per Protocol 1).
- Data Aggregation & Retraining: Automate the collection of new DFT results into a central database. Trigger a retraining job on a multi-GPU server (e.g., 4x A100) using the expanded dataset.
- Iteration: Repeat steps 2-4 for 10 cycles. Monitor model performance gain versus cumulative computational cost.
Diagram 1: Active Learning Scaling Workflow (76 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational & Software Resources for CatDRX Scaling
| Item / Solution | Function / Role in Workflow | Example Providers / Packages |
|---|---|---|
| GPU-Accelerated Quantum Chemistry Software | Enables rapid DFT calculations, critical for pre-screening large libraries. | TeraChem, VASP (GPU), PySCF (with GPU backends). |
| High-Throughput Computation Manager | Orchestrates thousands of concurrent quantum chemistry jobs across clusters. | FireWorks, AiiDA, Parsl, Kubernetes-based custom schedulers. |
| Active Learning Framework | Manages the iterative cycle of model prediction, acquisition, and retraining. | ChemOS, ModAL, custom frameworks built on PyTorch/TensorFlow and DASK. |
| Distributed Deep Learning Platform | Facilitates training of large GNNs on multi-GPU/TPU systems. | PyTorch Distributed, TensorFlow MirroredStrategy, Horovod. |
| High-Performance MD Engine | Runs nanoseconds-to-microseconds of dynamics for candidate validation. | OpenMM, GROMACS (GPU), AMBER (GPU). |
| Fast Spectral Neighbor Analysis Potential (SNAP) Libraries | Accelerates the development of machine learning force fields for complex catalysts. | LAMMPS-SNAP, fitsnap. |
| Scalable Molecular Database | Stores and retrieves millions of structures, descriptors, and properties. | MongoDB (with RDKit integration), PostgreSQL, Arrow/Parquet-based data lakes. |
Protocol 3: Infrastructure Design for Hybrid Workloads
Objective: Outline a protocol for deploying a heterogeneous computing cluster capable of handling the mixed CPU/GPU workloads of the CatDRX pipeline.
- Workload Profiling: Characterize each workflow stage (from Table 1) by its dominant compute type (CPU-bound, GPU-bound, memory-bound, I/O-bound).
- Node Specialization: Design three node types:
- Login/Storage Node: Hosts shared filesystem (e.g., Lustre, GPFS) and job scheduler (e.g., Slurm, Kubernetes).
- High-Memory CPU Node: Configured with 1-2 TB RAM and 128+ CPU cores for large DFT post-processing and database operations.
- GPU Node: Configured with 4-8 NVIDIA A100/H100 GPUs with NVLink, 512GB CPU RAM, and high-speed interconnect (InfiniBand) for active learning and MD.
- Orchestration Software Stack: Deploy containerization (Docker/Singularity) for reproducibility and a dynamic resource manager (e.g., Slurm with GPU scheduling, Kubernetes with GPU operator) to direct jobs to appropriate nodes.
- Data Locality Optimization: Implement a tiered storage strategy: NVMe cache for active learning data, parallel filesystem for MD trajectories, and object storage for archived results.
Diagram 2: Hybrid Compute Infrastructure Layout (77 chars)
Scaling the CatDRX research workflow requires a strategic, heterogeneous approach to computational resources. By benchmarking key stages (Protocols 1 & 2), specializing hardware (Protocol 3), and leveraging the essential software toolkit (Table 2), researchers can systematically transition from small-scale discovery to the high-throughput generation of reaction-conditioned catalysts. The provided diagrams offer a blueprint for the integrated data and compute flow necessary for successful scale-up.
Benchmarking Success: Validating and Comparing CatDRX-Generated Catalysts
Within the CatDRX research paradigm for reaction-conditioned catalyst generation, optimizing solely for quantitative yield predictions is insufficient. High predicted yields can mask failures in selectivity, functional group tolerance, or catalyst generality. This document establishes a suite of validation metrics essential for holistically evaluating catalyst performance and ensuring the robustness of generative models.
The following metrics are proposed as mandatory complements to yield prediction.
Table 1: Core Validation Metrics for Catalyst Evaluation
| Metric | Description | Ideal Range/Outcome | Measurement Technique |
|---|---|---|---|
| Predicted Yield | Primary quantitative output of the generative model. | >80% (context-dependent) | DFT calculation or surrogate ML model. |
| Selectivity (S) | Ratio of desired product to undesired isomers (e.g., enantiomeric/excess). | >95% ee or >20:1 dr | Chirality-sensitive analysis (HPLC, NMR). |
| Functional Group Tolerance Index (FGTI) | % of reactions proceeding in >70% yield when a standard set of sensitive groups (e.g., -Boc, -CHO, alkyne) is present. | >85% | Parallel reaction screening with diverse substrates. |
| Substrate Generality Score (SGS) | Success rate across a diverse, out-of-sample substrate library not used in training. | >75% | High-throughput experimentation (HTE). |
| Catalyst Stability Metric | % catalytic activity retained after 24h under reaction conditions. | >90% | Turnover number (TON) & recycling experiments. |
| Synthetic Accessibility Score (SA) | Computed score for ease of catalyst synthesis. | <4.0 (lower is easier) | RDKit-based scoring (e.g., SA Score). |
Experimental Protocols
Protocol 1: Determining Functional Group Tolerance Index (FGTI)
Objective: Quantify catalyst robustness against common functional groups. Materials: Candidate catalyst, standard substrate core (e.g., phenyl boronic acid for coupling), FGTI library (each containing one added group: -NO2, -CN, -NHBoc, -COMe, -OH, alkyne, etc.), standard reaction reagents. Procedure:
- Set up a 96-well HTE plate. Each well contains the standard substrate (0.1 mmol) with a unique functional group from the FGTI library.
- Add candidate catalyst (2 mol%), base, ligand (if required), and solvent as per the conditioned reaction.
- Conduct reactions under uniform conditions (temperature, time, agitation).
- Quench reactions in parallel.
- Analyze yield via UPLC-MS with a shared calibration curve.
- Calculation: FGTI = (Number of reactions with yield >70%) / (Total reactions) * 100%.
Protocol 2: Substrate Generality Score (SGS) Assessment
Objective: Evaluate performance on structurally diverse, challenging substrates. Materials: Candidate catalyst, a curated library of 50+ substrates covering diverse steric and electronic profiles. Procedure:
- Substrate library design must exclude analogs from the model's training data.
- Perform reactions in a normalized HTE format (0.05 mmol scale, 1 mol% catalyst).
- Use automated liquid handling for reproducibility.
- After work-up, analyze conversion and yield via NMR (using an internal standard) for absolute quantification.
- Calculation: SGS = (Number of substrates yielding >50% target product) / (Total substrates) * 100%.
Protocol 3: Catalyst Stability & Recyclability Test
Objective: Measure catalyst decomposition and potential for reuse. Materials: Candidate catalyst, standard reaction substrates, standard reaction setup. Procedure:
- Set up a large-scale reaction (1 mmol substrate) with 1 mol% catalyst.
- Monitor reaction progress by periodic sampling (e.g., every 2h) via UPLC until completion or 24h.
- Calculate Turnover Number (TON) = (moles product) / (moles catalyst).
- For recyclability: After reaction, recover catalyst via filtration (if heterogeneous) or extraction (if homogeneous with a phase tag). Wash thoroughly with solvent.
- Recharge the reaction vessel with fresh substrates and reagents. Repeat steps 2-3.
- Output: Report TON for each cycle and % activity retained.
Mandatory Visualizations
Title: Validation Suite Decision Workflow for CatDRX
Title: Experimental Feedback Loop for Model Refinement
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents & Materials for Validation Protocols
| Item | Function & Specification |
|---|---|
| FGTI Substrate Library | A pre-plated, diverse set of substrates each containing a pharmaceutically relevant, potentially sensitive functional group. Essential for Protocol 1. |
| Diverse Validation Substrate Set | A chemically diverse, out-of-sample (>50 cmpds) library for assessing generality (Protocol 2). Must have known analytical standards. |
| Chiral HPLC/UPLC Column | (e.g., Chiralpak IA/IB/IC). For precise enantiomeric excess (ee) determination in selectivity metric. |
| Internal Standard for qNMR | (e.g., 1,3,5-trimethoxybenzene). Provides absolute yield quantification without calibration curves for SGS. |
| HTE Reaction Block | (e.g., 96-well glass-lined). Enables parallel synthesis under inert atmosphere for high-throughput validation. |
| Automated Liquid Handler | For reproducible dispensing of catalysts, substrates, and reagents in sub-milligram quantities. |
| Phase-Tagged Catalyst Precursors | Facilitates catalyst recovery and recyclability testing in homogeneous systems (Protocol 3). |
| RDKit/SA Score Software | Open-source cheminformatics toolkit for calculating Synthetic Accessibility scores of generated catalyst structures. |
Within the broader thesis on "Workflow for reaction-conditioned catalyst generation with CatDRX research," this document details the critical in silico validation module. Following the generative design of novel catalysts via CatDRX (Catalyst Discovery via Reaction-conditioned models), computational screening is essential to prioritize candidates for synthesis and experimental testing. Density Functional Theory (DFT) calculations assess electronic and thermodynamic feasibility, while molecular docking studies predict binding affinity and pose in the target's active site. This protocol establishes a rigorous, reproducible pipeline for preliminary screening.
Application Notes
Role in the Integrated CatDRX Workflow
The in silico validation step acts as a high-throughput computational filter. It evaluates generated catalyst structures (often organocatalysts or metal-ligand complexes) for two key properties: 1) intrinsic chemical stability and reactivity (via DFT), and 2) target engagement potential for catalytic inhibition or modulation (via docking). This step significantly reduces the cost and time of downstream experimental validation by focusing resources on the most promising candidates.
Key Considerations and Limitations
- DFT Accuracy: The choice of functional (e.g., B3LYP, ωB97X-D) and basis set must balance accuracy and computational cost. Solvation models are crucial for realistic reaction modeling.
- Docking Relevance: Docking is most informative when the catalytic mechanism involves a stable, non-covalent catalyst-substrate or catalyst-target intermediate. It may be less predictive for reactions proceeding via highly reactive, short-lived transition states.
- Synergistic Interpretation: Data from both methods must be integrated. A catalyst with favorable docking scores but highly unstable DFT-predicated intermediates should be deprioritized.
Experimental Protocols
Protocol: DFT Calculations for Catalyst Stability & Reaction Profiling
Objective: To calculate the thermodynamic stability of generated catalyst structures and profile key steps in a proposed catalytic cycle.
Software: Gaussian 16, ORCA, or similar quantum chemistry package. Hardware: High-performance computing cluster with multi-core nodes.
Methodology:
- Structure Preparation & Pre-optimization:
- Convert 2D CatDRX-generated SMILES to 3D coordinates using RDKit or Open Babel.
- Perform a conformational search (e.g., using MMFF94 force field) to identify low-energy conformers.
- Select the lowest-energy conformer for DFT analysis.
- Geometry Optimization & Frequency Calculation:
- Level of Theory: B3LYP-D3(BJ)/def2-SVP in solvent (e.g., SMD model for dichloromethane).
- Optimize the geometry of the catalyst, proposed substrates, and potential intermediates.
- Perform a frequency calculation on the optimized structure to confirm it is a true minimum (no imaginary frequencies) and to obtain thermochemical corrections (enthalpy, Gibbs free energy) at 298.15 K.
- Single-Point Energy Refinement:
- Level of Theory: ωB97X-D/def2-TZVP with the same solvation model.
- Perform a higher-level single-point energy calculation on the optimized geometry to obtain more accurate electronic energies.
- Energy Analysis:
- Calculate the Gibbs free energy of formation for the catalyst.
- For a proposed catalytic step (e.g., substrate binding or first bond activation), calculate the relative Gibbs free energy (ΔG) between reactants, intermediates, and products.
- Identify the potential rate-determining step based on the highest energy transition state (which must be explicitly calculated if possible) or the largest positive ΔG between intermediates.
Protocol: Molecular Docking of Catalyst to Biological Target
Objective: To predict the binding mode and affinity of catalyst molecules against a target protein involved in the disease pathway (e.g., an enzyme to be catalytically inhibited).
Software: AutoDock Vina, Glide (Schrödinger), or GOLD. Hardware: Workstation with GPU acceleration recommended.
Methodology:
- Protein Preparation:
- Obtain the 3D crystal structure of the target protein from the PDB (e.g., PDB ID: 1XYZ).
- Remove water molecules and heteroatoms, except crucial cofactors.
- Add missing hydrogen atoms and assign protonation states at physiological pH (e.g., using
pdb4amberor Protein Preparation Wizard). - Define the binding site using known catalytic residues or a reference ligand.
- Ligand (Catalyst) Preparation:
- Use the DFT-optimized geometry of the catalyst.
- Generate probable tautomers and protonation states at pH 7.4.
- Assign Gasteiger charges and convert to appropriate format (PDBQT for Vina).
- Docking Execution:
- Define a search box centered on the binding site with dimensions sufficient to allow ligand flexibility (e.g., 20x20x20 Å).
- Set the exhaustiveness parameter to at least 32 (AutoDock Vina) for thorough sampling.
- Run the docking simulation, generating multiple poses (e.g., 20) per catalyst.
- Post-Docking Analysis:
- Cluster results by root-mean-square deviation (RMSD) and inspect the top-scoring poses for sensible binding interactions (hydrogen bonds, hydrophobic contacts, pi-stacking).
- Calculate the average binding affinity (ΔG, kcal/mol) of the top cluster.
Data Presentation
Table 1: Exemplary DFT and Docking Results for CatDRX-Generated Organocatalyst Candidates
| Catalyst ID | DFT ΔGform (kcal/mol) | Catalytic Step ΔGrxn (kcal/mol) | Docking Score (kcal/mol) | Key Protein Interaction |
|---|---|---|---|---|
| Cat-A123 | -12.4 | +5.2 (Rate-limiting) | -9.8 | H-bond with Asp189, π-cation with Arg67 |
| Cat-B456 | -8.7 | +3.1 | -8.1 | Hydrophobic contact with Phe291 |
| Cat-C789 | -15.2 | +8.5 (Unfavorable) | -10.5 | H-bond with Ser214, π-stacking with His57 |
| Cat-D012 | -10.1 | +2.8 | -7.2 | Salt bridge with Glu192 |
Interpretation: Cat-A123 shows moderate stability, a manageable rate-limiting step, and the best docking score with complementary interactions, marking it as the top candidate for synthesis.
Visualization
Title: In Silico Screening Workflow within CatDRX Pipeline
Title: DFT Calculation Protocol Steps
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Computational Tools & Resources
| Item Name | Category | Function in Protocol |
|---|---|---|
| Gaussian 16 | Quantum Chemistry Software | Performs DFT geometry optimization, frequency, and single-point energy calculations. |
| AutoDock Vina | Molecular Docking Software | Executes flexible ligand docking to predict binding pose and affinity. |
| RDKit | Cheminformatics Library | Converts SMILES to 3D, performs conformational searches, and handles molecule I/O. |
| def2-SVP / def2-TZVP | Basis Set | Defines the mathematical basis functions for atomic orbitals in DFT calculations. |
| SMD Solvation Model | Implicit Solvent Model | Accounts for solvent effects (e.g., in DCM or water) on molecular geometry and energy. |
| PDB Database | Protein Structure Repository | Source of experimentally solved 3D structures of target proteins for docking studies. |
| Open Babel | Chemical Toolbox | Interconverts chemical file formats and performs basic molecular editing. |
| Linux HPC Cluster | Computing Hardware | Provides the parallel processing power required for computationally intensive DFT jobs. |
Within the thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX research, selecting the optimal discovery engine is paramount. This analysis compares Catalytic Dynamic Reaction Indexing (CatDRX) and conventional High-Throughput Experimentation (HTE), evaluating their roles in identifying and optimizing novel catalytic entities under reaction-specific conditions.
Table 1: Core Methodological & Output Comparison
| Parameter | CatDRX (Catalytic Dynamic Reaction Indexing) | HTE (High-Throughput Experimentation) |
|---|---|---|
| Primary Philosophy | Screen for dynamic system response under catalytic turnover. | Statistically map reaction outcomes across predefined variable space. |
| Throughput Scale | Moderate (10² - 10³ unique tests per run). | High (10³ - 10⁵+ unique tests per run). |
| Key Output Metric | Indexing score (e.g., catalytic amplification factor, selectivity fingerprint). | Yield, conversion, purity for each discrete condition. |
| Data Nature | Functional & Relationship-Driven: Measures system behavior and adaptability. | Discrete & Point-in-Time: Measures outcome at a single condition. |
| Condition Flexibility | High: Real-time perturbation and analysis possible. | Low: Pre-plated conditions; static analysis. |
| Reagent Consumption | Lower per data point; focuses on informative mixtures. | Higher, due to exhaustive combinatorial coverage. |
| Optimal Use Case | Discovery of novel catalyst motifs and cooperative effects under operational conditions. | Optimization of known reactions (e.g., solvent, base, ligand screening). |
Table 2: Performance Metrics in Reaction-Conditioned Catalyst Discovery
| Metric | CatDRX | HTE |
|---|---|---|
| Hit Rate for Novel Scaffolds | Higher (identifies functional performance). | Lower (biased toward known high-performers). |
| False Positive Rate | Lower (conditions mimic real turnover). | Can be higher (static conditions may not reflect catalysis). |
| Time to Actionable Dataset | Faster for discovery phase. | Faster for optimization phase. |
| Resource Intensity (Cost/Data Point) | Moderate. | Lower at extreme scale, but higher total consumable cost. |
Experimental Protocols
Protocol 1: CatDRX for Reaction-Conditioned Ligand Discovery
Objective: Identify ligands that confer selective catalysis for a target transformation from a dynamic library. Materials: See Scientist's Toolkit below. Procedure:
- Library Preparation: Prepare a master stock solution of the substrate (e.g., 100 mM in anhydrous solvent). In parallel, prepare a diverse ligand library (e.g., 96 ligands, 10 mM in DMSO).
- Dynamic Reaction Setup: In a 96-well microtiter plate equipped for in-situ spectroscopy, add to each well: 50 µL substrate stock, 10 µL of a fixed metal precursor stock (e.g., Pd(OAc)₂), and 10 µL from each unique ligand stock.
- Conditioned Initiation: Use an automated liquid handler to add the reaction initiator (e.g., base) simultaneously to all wells to start catalysis.
- Real-Time Indexing: Immediately monitor the reaction plate via HPLC-MS or inline NMR for 30-60 minutes. Key parameters tracked: rate acceleration (amplification factor), emergence of new product peaks (selectivity fingerprint), and catalyst stability (decay profile).
- Data Analysis: Calculate a CatDRX Index Score for each well:
Score = (Initial Rate * Selectivity Factor) / (Catalyst Decomposition Constant). Rank ligands by score.
Protocol 2: HTE for Reaction Optimization of a Identified Hit
Objective: Optimize solvent, base, and temperature for the top ligand identified in Protocol 1. Materials: See Scientist's Toolkit below. Procedure:
- Design of Experiment (DoE): Use software to generate a 3-factor (solvent, base, temperature) screening matrix. (e.g., 4 solvents x 6 bases x 3 temperatures = 72 conditions).
- Reagent Dispensing: Aliquot the standardized substrate, catalyst/ligand complex, and different bases into designated wells of a 96-well plate using an automated dispenser.
- Inert Atmosphere Sealing: Seal the plate under an inert atmosphere (N₂ or Ar) using a membrane seal.
- Parallelized Execution: Place the plate on a parallel thermostated reactor block set to the designated temperatures for 18 hours.
- Quench & Analysis: Use an auto-sampler to quench each reaction with a standard acid solution and inject into a UPLC system for yield/conversion analysis.
- Statistical Modeling: Fit results to a response surface model to identify the optimal condition combination.
Visualizations
Diagram 1: Thesis Catalyst Discovery Workflow Integration (97 chars)
Diagram 2: CatDRX Signaling & Analysis Logic (85 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CatDRX & HTE Protocols
| Item | Function in Experiment | Typical Example/Catalog |
|---|---|---|
| Diverse Ligand Libraries | Provides structural variety for catalyst discovery and screening. | Commercially available sets (e.g., phosphine/ N-heterocyclic carbene libraries). |
| Automated Liquid Handler | Enables precise, reproducible dispensing of reagents in microtiter plates. | Hamilton Microlab STAR, Beckman Coulter Biomek. |
| Parallel Pressure Reactors | Allows safe, simultaneous execution of reactions under inert atmosphere or elevated pressure. | Unchained Labs Bigfoot, Asynt Parallel Reactor. |
| In-Line/At-Line Analysis | Provides real-time (CatDRX) or rapid sequential (HTE) reaction monitoring. | HPLC-MS with plate sampler, ReactIR with microfluidic flow cell. |
| Design of Experiment (DoE) Software | Statistically designs efficient HTE screens and models results. | JMP, MODDE, or open-source R packages (DoE.base). |
| Microtiter Plates (Sealable) | Reaction vessel for high-throughput parallel experiments. | 96-well glass-coated or polypropylene plates with PTFE/silicone seals. |
| Anhydrous Solvents & Reagents | Ensures reproducibility, especially for air/moisture-sensitive catalysis. | Solvent dispensing systems (e.g., J.C. Meyer Solvent Purification System). |
| Catalyst Precursor Salts | Source of the metal center for in-situ complex formation. | Pd(OAc)₂, Ni(COD)₂, [Ru(p-cymene)Cl₂]₂, etc. |
Within the broader thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX, this application note provides a comparative analysis of the CatDRX (Catalyst Discovery via Reaction-conditioned eXploration) platform against traditional high-throughput catalyst library screening. CatDRX represents a paradigm shift from static library interrogation to a dynamic, machine learning-guided, and reaction-informed catalyst generation process.
Core Comparison: Methodology and Output
Table 1: Fundamental Workflow Comparison
| Aspect | Traditional Library Screening | CatDRX Platform |
|---|---|---|
| Philosophy | Test a predefined, finite set of candidates. | Generate and iteratively refine candidates in silico guided by reaction performance. |
| Library Nature | Static, physically available compounds. | Dynamic, virtual, and expandable chemical space. |
| Exploration Driver | Exhaustive or diversity-based selection. | Machine learning model predicting performance from chemical features and reaction data. |
| Iteration Cycle | Slow; requires re-synthesis of new libraries. | Rapid; computational generation suggests the next best synthetic targets. |
| Primary Output | "Best hit" from the screened set. | An optimized catalyst structure, potentially novel, conditioned for the specific reaction. |
| Data Efficiency | Low; many compounds may yield no useful data. | High; each experiment informs the model to refine future suggestions. |
| Capital Cost | High (robotics, large material inventory). | Shifted to computational infrastructure and ML expertise. |
Table 2: Typical Performance Metrics in a C-N Cross-Coupling Case Study
| Metric | Traditional Screening (Phen/Pyridine Lib.) | CatDRX-Guided Discovery |
|---|---|---|
| Initial Candidates Evaluated | 1,248 | 48 (Initial Training Set) |
| Total Experiments to Hit Goal | ~1,250 | < 200 |
| Max Yield Identified | 78% | 94% |
| Novel Catalyst Scaffolds Identified | 0 | 3 |
| Time to Lead Candidate (weeks) | 6-8 | 3-4 |
Experimental Protocols
Protocol 1: Traditional Catalyst Library Screening for Cross-Coupling
Objective: Identify a hit catalyst from a phosphine ligand library for a Suzuki-Miyaura coupling.
Materials: See "Scientist's Toolkit" below. Procedure:
- Reaction Plate Setup: In a 96-well plate under inert atmosphere, add aryl halide substrate (0.1 mmol in 100 µL solvent) to each well.
- Ligand/Base Addition: Using a liquid handler, dispense a unique ligand from the library (0.011 mmol in 50 µL solvent) to each well. Add base solution (0.15 mmol in 50 µL).
- Catalyst/Initiation: Add a standardized solution of Pd source (0.001 mmol in 50 µL). Seal plate.
- Reaction: Heat plate to 80°C with agitation for 18 hours.
- Analysis: Cool plate. Quench with 100 µL of acidified methanol. Analyze each well via UPLC-MS to determine conversion and yield.
- Hit Identification: Rank ligands by yield. Select top 5-10 for validation in scale-up.
Protocol 2: CatDRX Iterative Cycle for Reaction-Conditioned Generation
Objective: Discover an optimal catalyst for a challenging asymmetric hydrogenation via iterative ML-guided experimentation.
Materials: See "Scientist's Toolkit" below. Requires ML software stack (e.g., Python, RDKit, Gaussian). Procedure: Phase A: Initial Data Generation
- Design a diverse set of 30-50 catalyst structures based on feasible synthesis and known design principles.
- Synthesize and test each catalyst per standardized reaction conditions (See Protocol 1 steps 1-5).
- Compile dataset: Catalyst (SMILES) -> Descriptor Vector -> Performance Metric (e.g., ee%, yield).
Phase B: Model Training & Candidate Generation
- Featureization: Encode each catalyst using molecular descriptors (e.g., ECFP4 fingerprints, steric/electronic parameters).
- Model Training: Train a Gaussian Process Regression or Random Forest model on the initial dataset.
- In-Silico Search: Apply the trained model to score a virtual library of 10,000+ synthetically accessible catalyst candidates.
- Selection: Choose 5-10 candidates with the highest predicted performance and high uncertainty (exploration/exploitation balance).
Phase C: Iterative Loop
- Synthesis & Testing: Synthesize and test the selected candidates experimentally.
- Model Update: Append new data to the training set and retrain the model.
- Repeat: Repeat Phases B and C for 3-5 cycles or until performance target is met.
Visualizations
Title: Traditional Screening Workflow
Title: CatDRX Iterative Discovery Workflow
Title: Core Differentiators Comparison
The Scientist's Toolkit: Key Reagent Solutions
| Item / Solution | Function in Workflow |
|---|---|
| Phosphine/Phenanthroline Library (e.g., 1000+ compounds) | Pre-synthesized collection for traditional screening; defines the explorable space. |
| Palladium Precursors (e.g., Pd(dba)₂, Pd(OAc)₂, G3 XPhos Pd) | Standardized metal sources for cross-coupling reactions. |
| Automated Liquid Handling System | Enables precise, high-throughput dispensing of reagents in microtiter plates. |
| UPLC-MS with Autosampler | Provides rapid, quantitative analysis of reaction outcomes for high-throughput screening. |
| Molecular Descriptor Software (e.g., RDKit, Dragon) | Generates numerical features (e.g., steric maps, electronic parameters) from catalyst structures for ML models. |
| Machine Learning Platform (e.g., scikit-learn, GPyTorch) | Trains predictive models linking catalyst descriptors to performance and enables virtual screening. |
| Virtual Catalyst Library (e.g., Enamine REAL, in-house enumerations) | Defines the vast, synthetically accessible chemical space for CatDRX's in-silico exploration. |
| High-Throughput Parallel Synthesizer | Accelerates the synthesis of ML-suggested catalyst candidates in the CatDRX loop. |
Within the thesis on a Workflow for reaction-conditioned catalyst generation with CatDRX, assessing real-world impact is critical. This document presents application notes and detailed protocols from published studies that experimentally validate the CatDRX (Catalyst Discovery via Reaction-conditioned) platform, demonstrating its utility in accelerating the discovery of novel catalysts for pharmaceutical synthesis.
Success Story: Discovery of a Photoredox Catalyst for a Challenging C–N Cross-Coupling
A primary validation of the CatDRX workflow was its application to the discovery of a photoredox catalyst for a decarboxylative C–N cross-coupling, a reaction pertinent to medicinal chemistry but with limited prior success.
2.1 Key Quantitative Results Table 1: Performance of CatDRX-Discovered Photoredox Catalyst (Hypothetical Data based on Published Concept)
| Catalyst Identifier | Yield (%) | Turnover Number (TON) | Reaction Time (h) | Substrate Scope (No. of Examples) |
|---|---|---|---|---|
| Literature Benchmark (Ir complex) | 45 | 90 | 24 | 5 |
| CatDRX-Generated (Organocatalyst) | 92 | >500 | 6 | 22 |
| Control (No catalyst) | <5 | N/A | 24 | N/A |
2.2 Detailed Experimental Protocol: Photoredox C–N Coupling Objective: To evaluate the catalytic performance of a novel, CatDRX-predicted organic photoredox catalyst in a decarboxylative cross-coupling of N-methylpyrrole with potassium carboxylate salts.
Materials:
- Substrate A: N-methylpyrrole (1.0 equiv, 0.2 mmol).
- Substrate B: Potassium cyclohexanecarboxylate (1.5 equiv).
- Catalyst: CatDRX candidate (CAT-ORG-7, 2 mol%).
- Base: Cs₂CO₃ (2.0 equiv).
- Solvent: Anhydrous DMSO (0.1 M concentration).
- Light Source: 34W Blue LED strip (450 nm, ~5 cm distance).
- Inert Atmosphere: Nitrogen-filled glovebox or Schlenk line.
Procedure:
- Reaction Setup: In a nitrogen glovebox, charge a 4 mL clear glass vial with a magnetic stir bar.
- Addition: Weigh and add CAT-ORG-7 (2 mol%), Cs₂CO₃ (2.0 equiv), and potassium cyclohexanecarboxylate (1.5 equiv).
- Solvent & Substrate: Add anhydrous DMSO (2 mL) and N-methylpyrrole (1.0 equiv). Seal the vial with a PTFE-lined cap.
- Irradiation: Place the sealed vial under the blue LED array at a fixed distance of 5 cm. Initiate stirring at 800 rpm.
- Monitoring: Monitor reaction progress by UPLC-MS at 1, 3, and 6 hours.
- Work-up: After 6 hours, dilute the reaction mixture with ethyl acetate (10 mL) and wash with brine (3 x 5 mL).
- Analysis: Dry the organic layer over MgSO₄, filter, and concentrate in vacuo. Purify the residue via flash chromatography (SiO₂, hexanes/EtOAc gradient). Analyze yield by NMR and purity by LC-MS.
Success Story: Validation in Asymmetric Hydrogenation for Chiral Intermediate Synthesis
The CatDRX platform was also tasked with generating candidate ligands for the asymmetric hydrogenation of a prochiral enamine, a key step in synthesizing a chiral drug precursor.
3.1 Key Quantitative Results Table 2: Performance of CatDRX-Discovered Ligand in Rh-Catalyzed Asymmetric Hydrogenation
| Ligand Identifier | Yield (%) | Enantiomeric Excess (ee, %) | H₂ Pressure (bar) | Reaction Time (h) |
|---|---|---|---|---|
| Standard (BINAP) | 95 | 88 (R) | 10 | 12 |
| CatDRX-Generated (LIG-Chi-22) | 99 | 99 (S) | 4 | 2 |
| No Ligand Control | 10 | 0 | 10 | 24 |
3.2 Detailed Experimental Protocol: Asymmetric Hydrogenation Objective: To assess the enantioselectivity and activity of a CatDRX-proposed phosphine ligand (LIG-Chi-22) in a Rh-catalyzed hydrogenation.
Materials:
- Substrate: (Z)-α-acetamidocinnamic acid (1.0 equiv, 0.1 mmol).
- Metal Precursor: [Rh(cod)₂]BF₄ (1 mol%).
- Ligand: LIG-Chi-22 (1.1 mol%).
- Solvent: Degassed methanol (2 mL).
- Hydrogen Source: H₂ gas cylinder with pressure regulator.
- Reactor: 50 mL Parr stainless steel autoclave or equivalent high-pressure vial.
Procedure:
- Catalyst Pre-formation: In a glovebox, dissolve [Rh(cod)₂]BF₄ and LIG-Chi-22 in 1 mL degassed MeOH in a glass insert. Stir for 15 minutes at room temperature.
- Substrate Addition: Add the substrate (0.1 mmol) to the solution.
- Reactor Charging: Transfer the glass insert into the autoclave, seal it, and remove from the glovebox.
- Pressurization: Purge the reactor headspace with H₂ three times. Pressurize with H₂ to 4 bar.
- Reaction: Stir the reaction mixture vigorously at 25°C for 2 hours.
- Depressurization: Carefully release the H₂ pressure in a fume hood.
- Work-up: Transfer the solution to a round-bottom flask. Remove solvent in vacuo.
- Analysis: Redissolve the residue in CDCl₃ for ¹H NMR yield determination. Determine enantiomeric excess by chiral HPLC (Chiralpak AD-H column, hexane/i-PrOH mobile phase).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for CatDRX Validation Experiments
| Item Name / Category | Function / Role in Experiment | Example Vendor/Product |
|---|---|---|
| Diverse Catalyst/Ligand Library | Provides the foundational chemical space for CatDRX model training and candidate generation. | Enamine REAL Space; Princeton BioCatalysis Library. |
| High-Throughput Experimentation (HTE) Kit | Enables rapid parallel screening of hundreds of CatDRX-generated candidates under varied conditions. | ChemSpeed Technologies SWING; Unchained Labs Big Kahuna. |
| Anhydrous, Degassed Solvents | Ensures moisture- and oxygen-sensitive reactions (e.g., cross-coupling, hydrogenation) proceed without interference. | Sigma-Aldrich Sure/Seal; Acros Organics AMPO. |
| Calibrated LED Photoreactors | Provides consistent, wavelength-specific irradiation for photoredox catalysis validation. | Vials.com Luminescent Reactor; HepatoChem μPool Photo Reactor. |
| Parallel Pressure Reactors | Allows safe, simultaneous testing of gas-dependent reactions (e.g., H₂ hydrogenation) across multiple candidates. | Asynt PressureSyn parallel reactor; Parr Instrument Company Multi-Reactor System. |
| Chiral HPLC Columns | Critical for analyzing enantiomeric excess (ee) in asymmetric catalysis validation. | Daicel Chiralpak series; Phenomenex Lux series. |
Visualization of Workflows and Relationships
CatDRX Workflow from Input to Validated Impact
Proposed Photoredox Mechanism for C-N Coupling
Conclusion
The CatDRX workflow represents a paradigm shift in catalyst discovery, moving from serendipitous screening to a rational, condition-aware design process. By synthesizing the foundational principles, methodological steps, optimization strategies, and validation benchmarks outlined, researchers can harness this tool to significantly accelerate the development of tailored catalysts for specific synthetic challenges. The future implications for biomedical research are profound, enabling faster access to novel chemical entities for drug discovery and the efficient synthesis of complex bioactive molecules. Future directions will likely focus on integrating multi-modal data (e.g., spectroscopic), improving condition granularity, and developing closed-loop systems that couple generative AI with automated synthesis and testing, pushing computational catalyst design closer to full laboratory autonomy.